

CD37 is a safe chimeric antigen receptor target to treat acute myeloid leukemia
- PMID: 38754420
- PMCID: PMC11228397
- DOI: 10.1016/j.xcrm.2024.101572
CD37 is a safe chimeric antigen receptor target to treat acute myeloid leukemia
Abstract
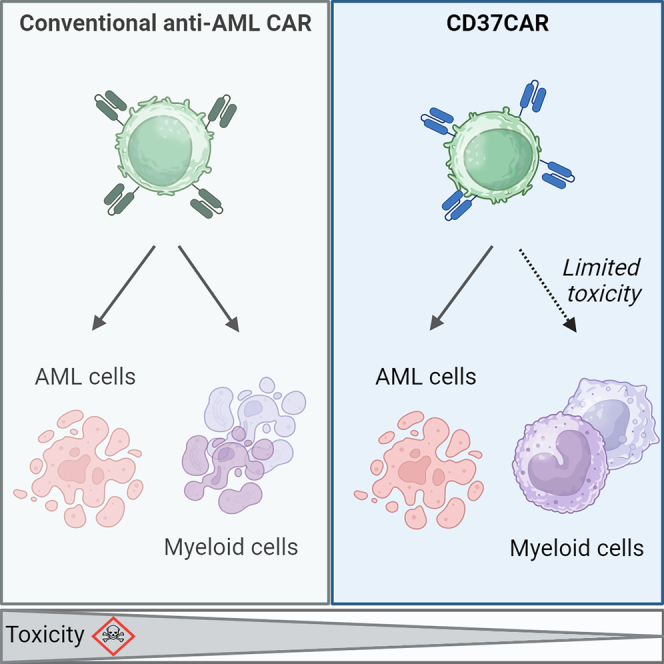
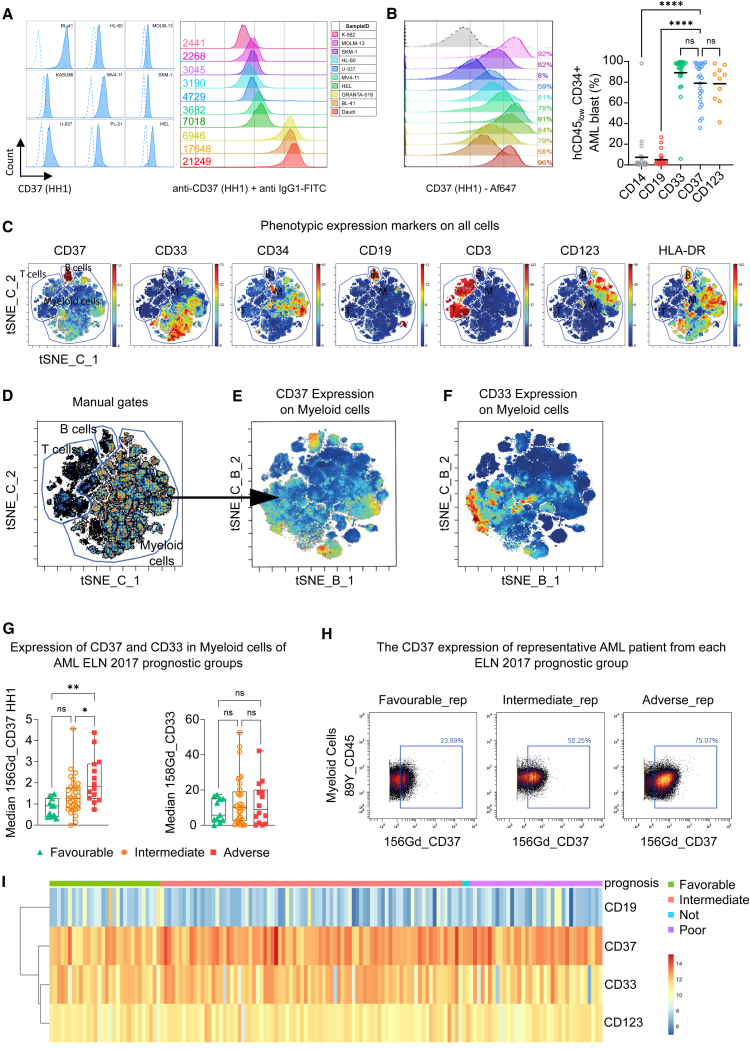
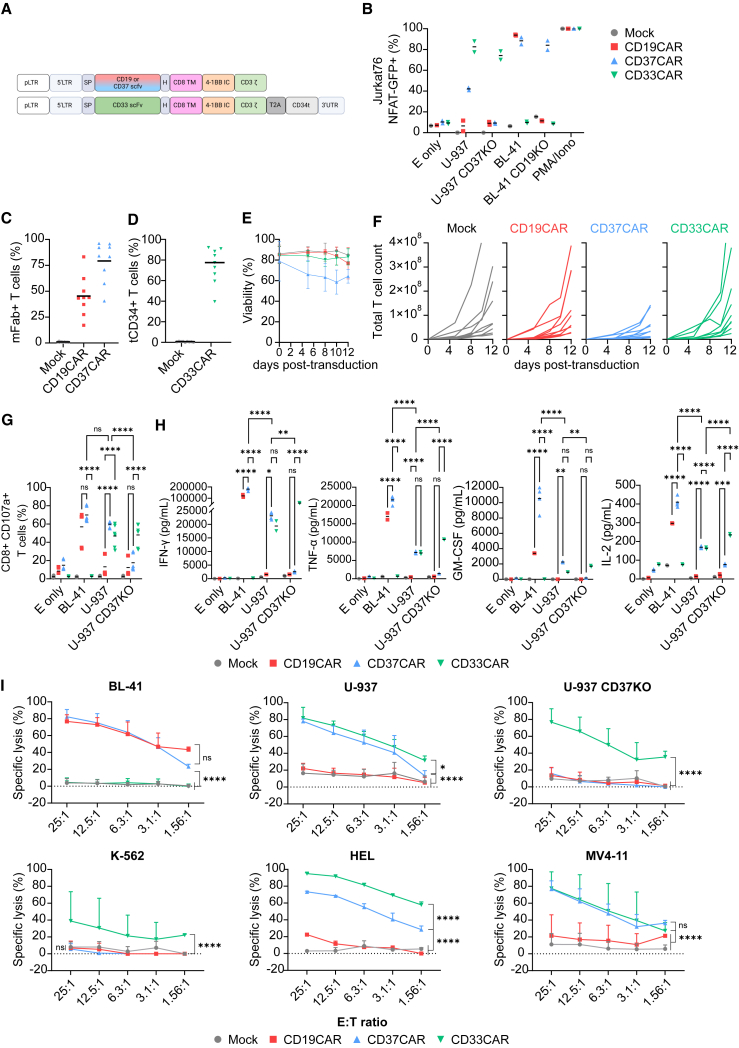
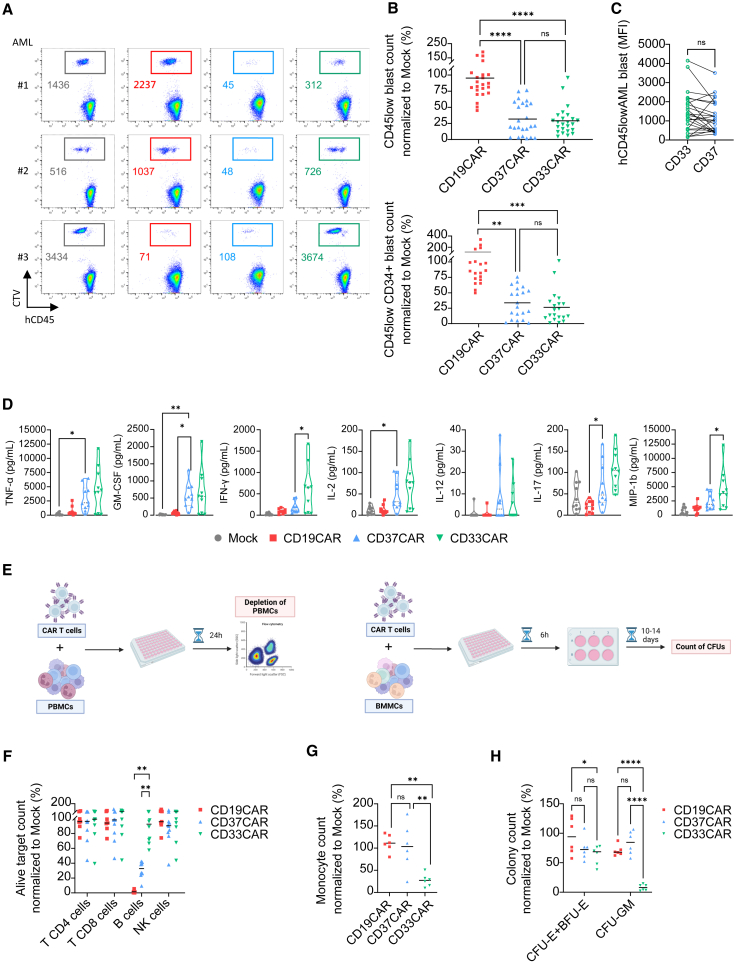
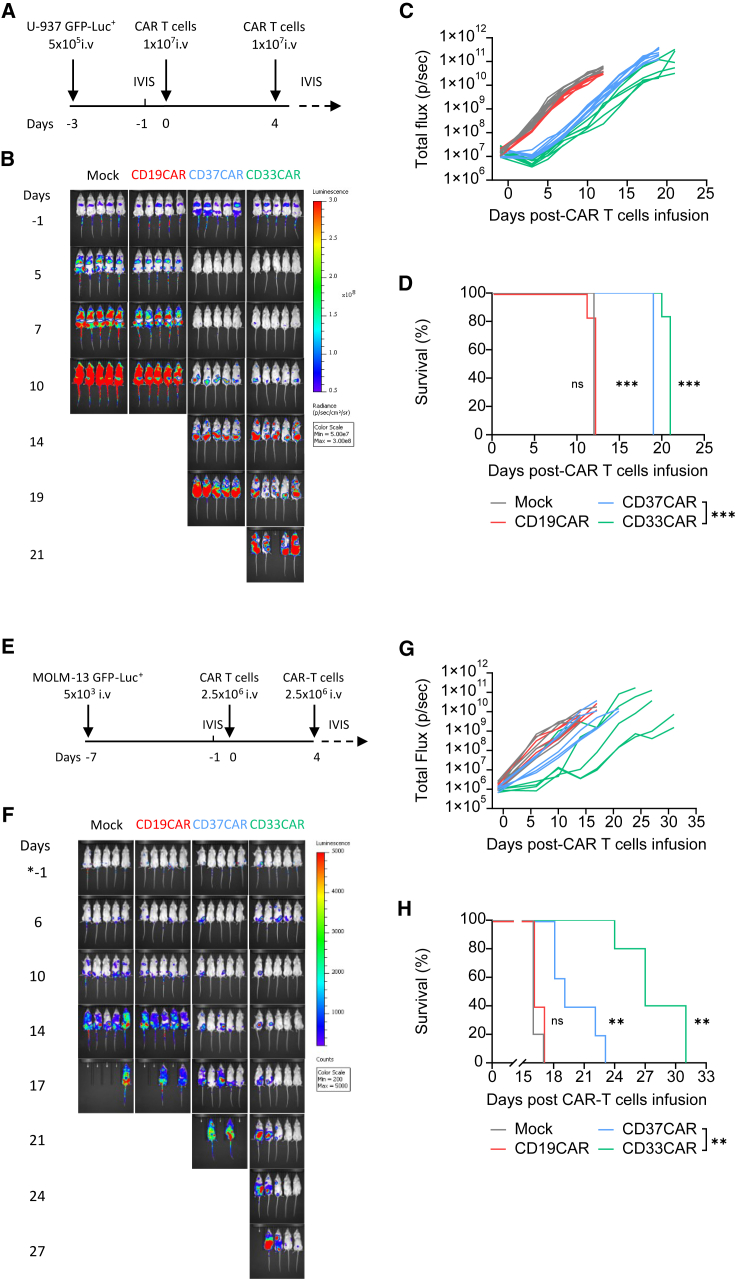
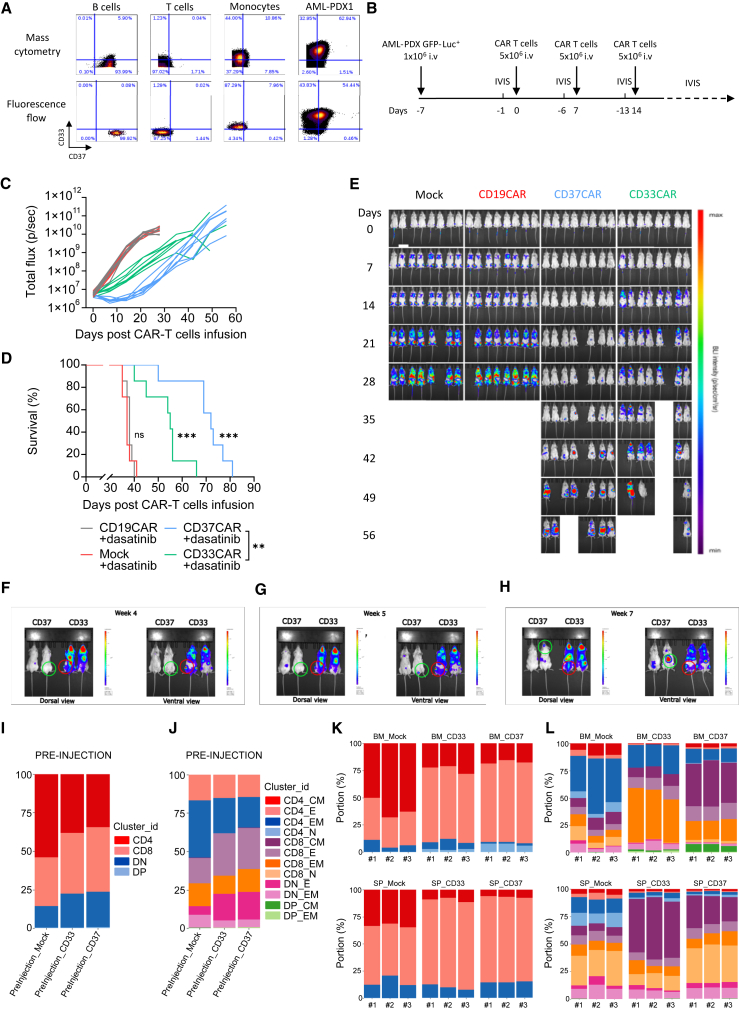
Acute myeloid leukemia (AML) is characterized by the accumulation of immature myeloid cells in the bone marrow and the peripheral blood. Nearly half of the AML patients relapse after standard induction therapy, and new forms of therapy are urgently needed. Chimeric antigen receptor (CAR) T therapy has so far not been successful in AML due to lack of efficacy and safety. Indeed, the most attractive antigen targets are stem cell markers such as CD33 or CD123. We demonstrate that CD37, a mature B cell marker, is expressed in AML samples, and its presence correlates with the European LeukemiaNet (ELN) 2017 risk stratification. We repurpose the anti-lymphoma CD37CAR for the treatment of AML and show that CD37CAR T cells specifically kill AML cells, secrete proinflammatory cytokines, and control cancer progression in vivo. Importantly, CD37CAR T cells display no toxicity toward hematopoietic stem cells. Thus, CD37 is a promising and safe CAR T cell AML target.
Keywords: AML; CAR T cell; CD37; acute myeloid leukemia; chimeric antigen receptor; hematopoietic stem cell; immunotherapy; patient-derived xenograft.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The CD37CAR construct has been patented (WO2017118745A1) and E.M.I., G.K., and S.W. are listed among the inventors. S.K. has received honoraria from TCR2 Inc., Miltenyi, Novartis, BMS, and GSK. S.K. is inventor of several patents in the field of immuno-oncology. S.K. received license fees from TCR2 Inc. and Carina Biotech. S.K. received research support from TCR2 Inc., Plectonic GmBH, Tabby Therapeutics, and Arcus Bioscience for work unrelated to this manuscript. The funding agencies had no role in the conduction and management of the presented research and were not involved in the preparation of this manuscript.
Figures
References
-
- Dohner H., Wei A.H., Appelbaum F.R., Craddock C., DiNardo C.D., Dombret H., Ebert B.L., Fenaux P., Godley L.A., Hasserjian R.P., et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140:1345–1377. doi: 10.1182/blood.2022016867. - DOI - PubMed
-
- Döhner H., Estey E., Grimwade D., Amadori S., Appelbaum F.R., Büchner T., Dombret H., Ebert B.L., Fenaux P., Larson R.A., et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447. doi: 10.1182/blood-2016-08-733196. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
