

ADAM9 promotes type I interferon-mediated innate immunity during encephalomyocarditis virus infection
- PMID: 38755212
- PMCID: PMC11098812
- DOI: 10.1038/s41467-024-48524-6
ADAM9 promotes type I interferon-mediated innate immunity during encephalomyocarditis virus infection
Abstract
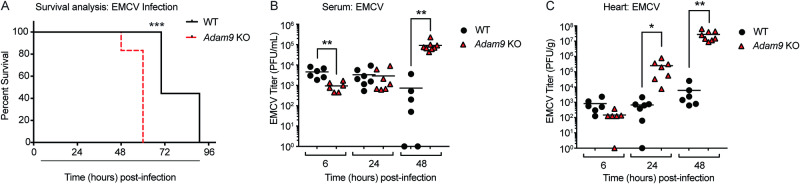
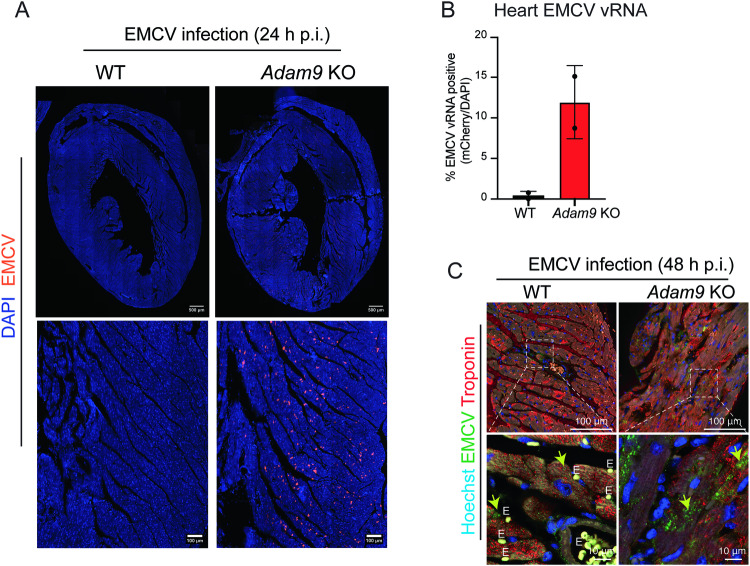
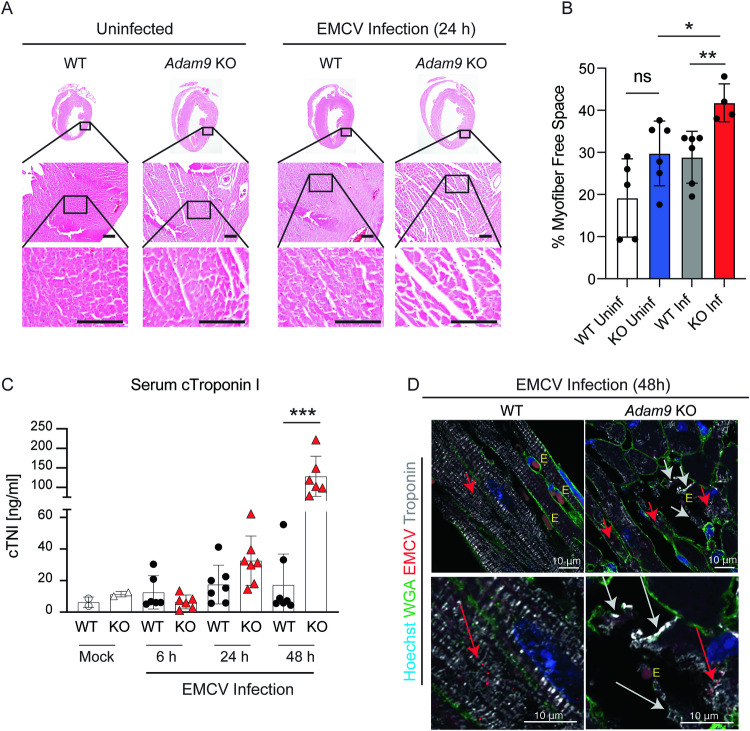
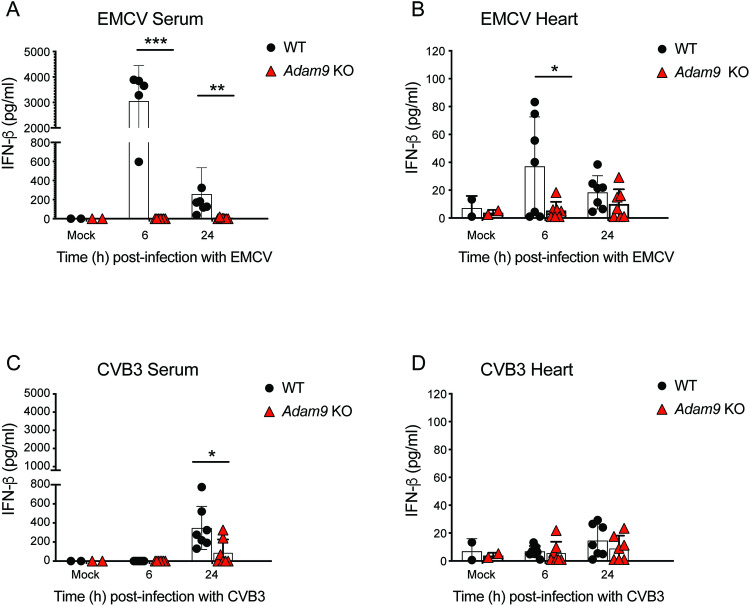
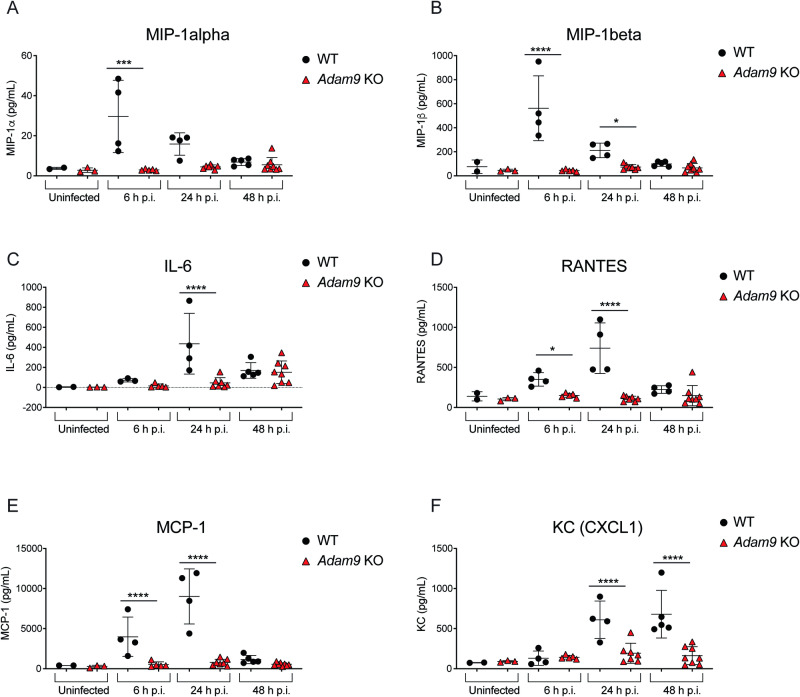
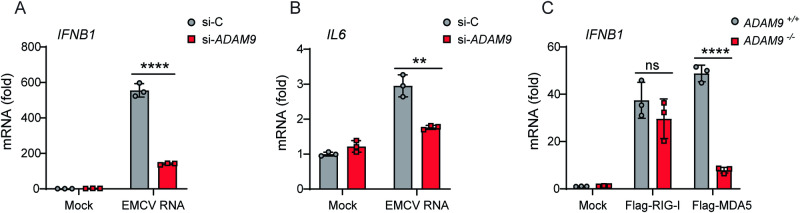
Viral myocarditis, an inflammatory disease of the heart, causes significant morbidity and mortality. Type I interferon (IFN)-mediated antiviral responses protect against myocarditis, but the mechanisms are poorly understood. We previously identified A Disintegrin And Metalloproteinase domain 9 (ADAM9) as an important factor in viral pathogenesis. ADAM9 is implicated in a range of human diseases, including inflammatory diseases; however, its role in viral infection is unknown. Here, we demonstrate that mice lacking ADAM9 are more susceptible to encephalomyocarditis virus (EMCV)-induced death and fail to mount a characteristic type I IFN response. This defect in type I IFN induction is specific to positive-sense, single-stranded RNA (+ ssRNA) viruses and involves melanoma differentiation-associated protein 5 (MDA5)-a key receptor for +ssRNA viruses. Mechanistically, ADAM9 binds to MDA5 and promotes its oligomerization and thereby downstream mitochondrial antiviral-signaling protein (MAVS) activation in response to EMCV RNA stimulation. Our findings identify a role for ADAM9 in the innate antiviral response, specifically MDA5-mediated IFN production, which protects against virus-induced cardiac damage, and provide a potential therapeutic target for treatment of viral myocarditis.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
