

SMAD2/3-SMYD2 and developmental transcription factors cooperate with cell-cycle inhibitors to guide tissue formation
- PMID: 38758030
- PMCID: PMC12053477
- DOI: 10.1093/procel/pwae031
SMAD2/3-SMYD2 and developmental transcription factors cooperate with cell-cycle inhibitors to guide tissue formation
Erratum in
-
Correction to: the following articles.Protein Cell. 2025 Aug 25:pwaf065. doi: 10.1093/procel/pwaf065. Online ahead of print. Protein Cell. 2025. PMID: 40853189 No abstract available.
Abstract
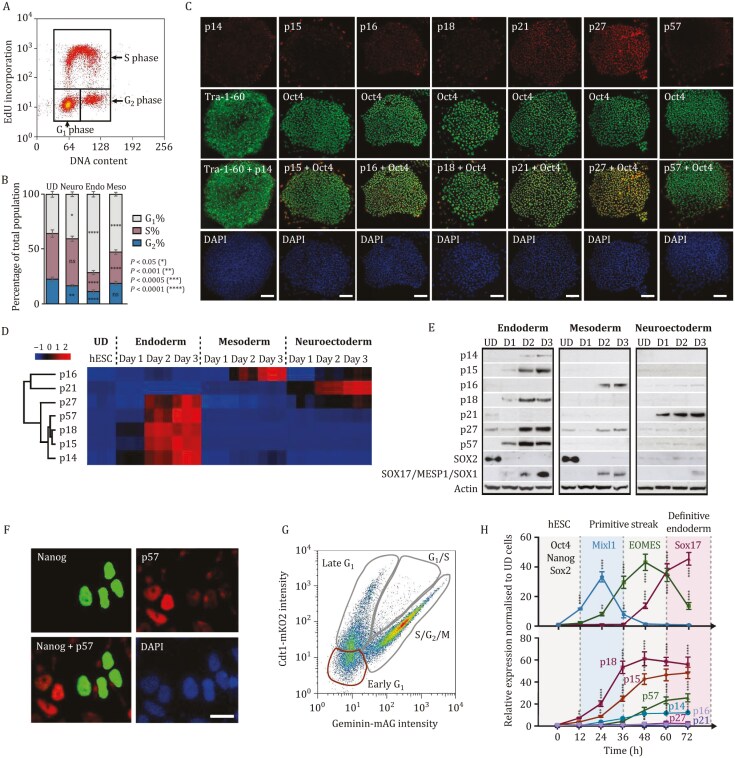
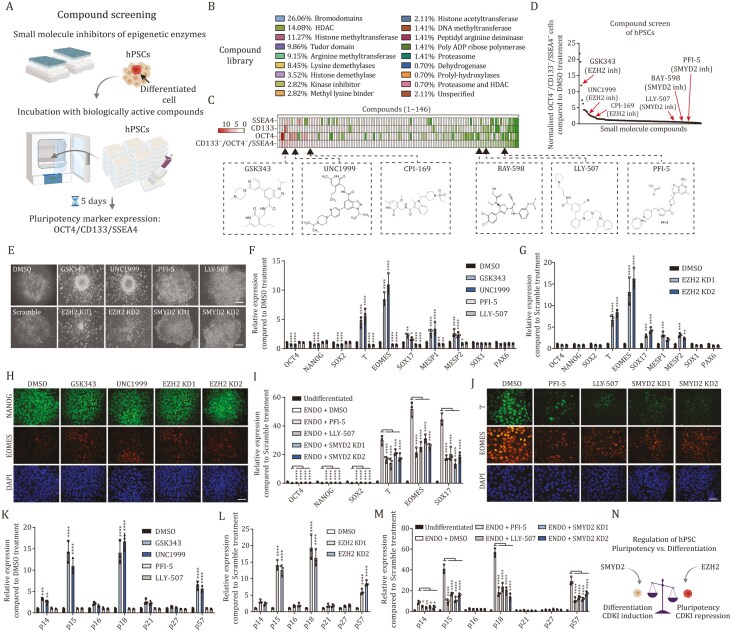
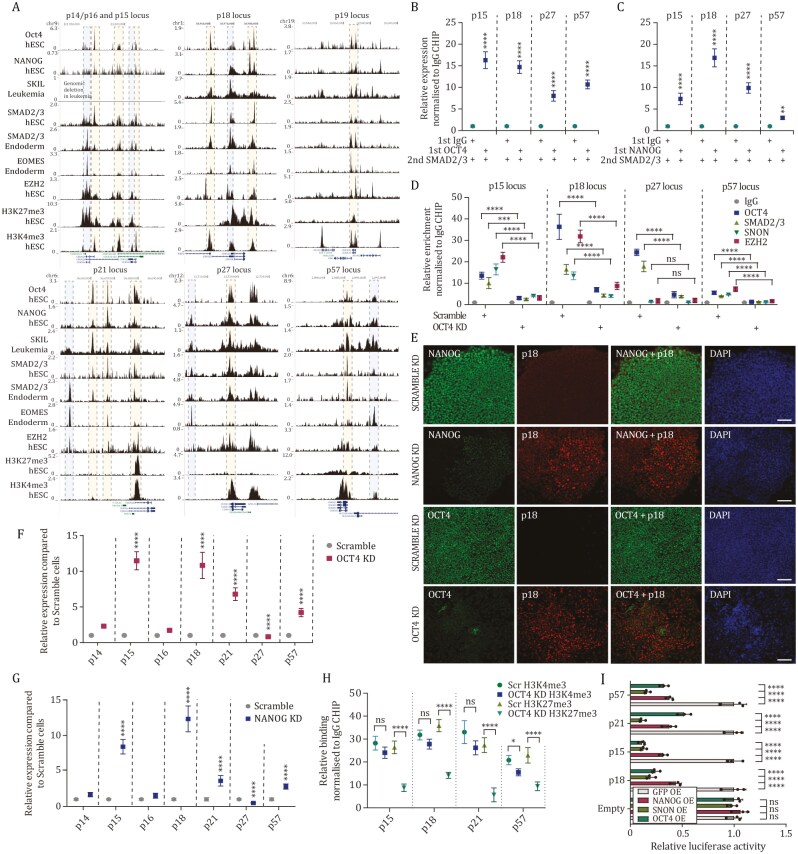
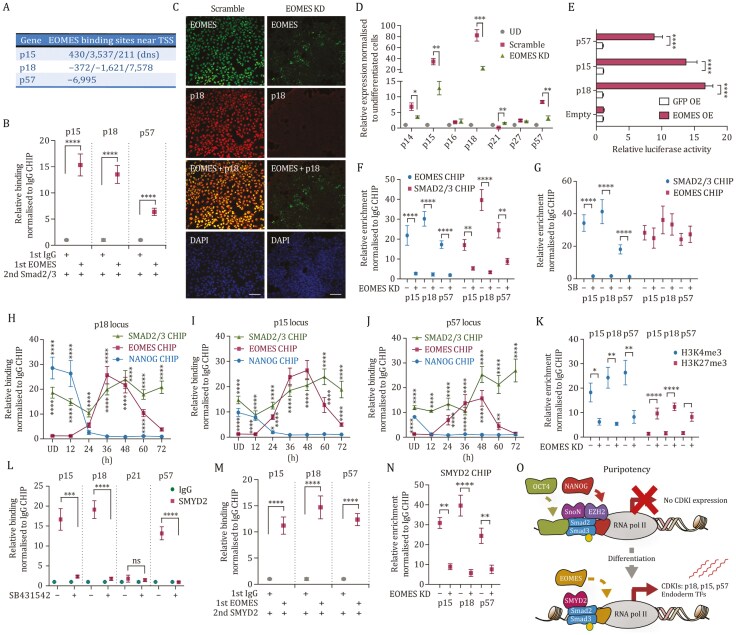
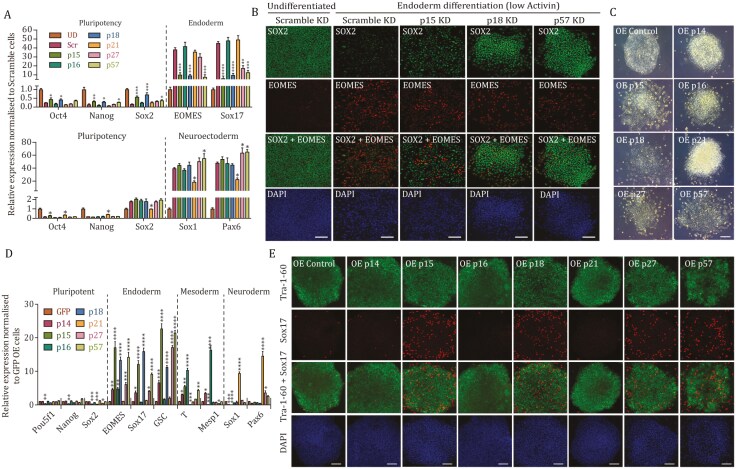
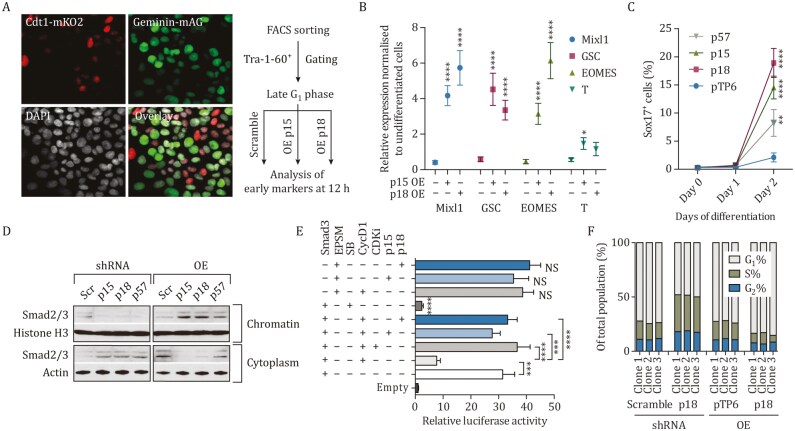
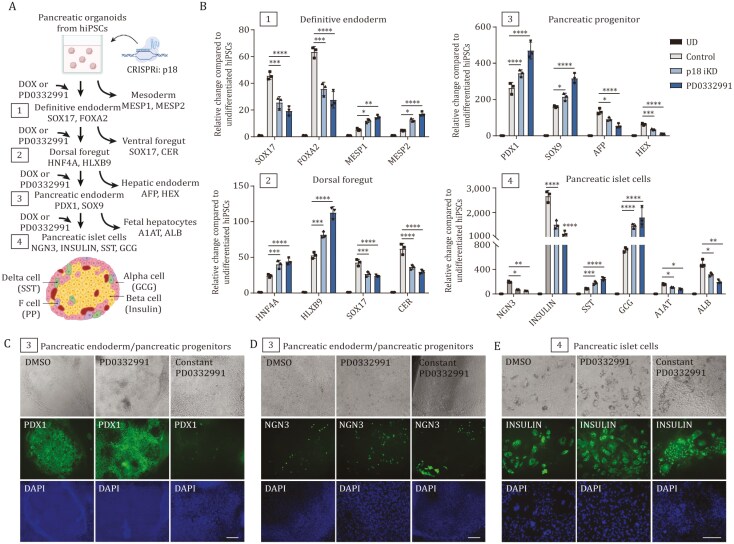
Tissue formation and organ homeostasis are achieved by precise coordination of proliferation and differentiation of stem cells and progenitors. While deregulation of these processes can result in degenerative disease or cancer, their molecular interplays remain unclear. Here we show that the switch of human pluripotent stem cell (hPSC) self-renewal to differentiation is associated with the induction of distinct cyclin-dependent kinase inhibitors (CDKIs). In hPSCs, Activin/Nodal/TGFβ signaling maintains CDKIs in a poised state via SMAD2/3-NANOG-OCT4-EZH2-SNON transcriptional complex. Upon gradual differentiation, CDKIs are induced by successive transcriptional complexes between SMAD2/3-SMYD2 and developmental regulators such as EOMES, thereby lengthening the G1 phase. This, in turn, induces SMAD2/3 transcriptional activity by blocking its linker phosphorylation. Such SMAD2/3-CDKI positive feedback loops drive the exit from pluripotency and stepwise cell-fate specification that could be harnessed for producing cells for therapeutic applications. Our study uncovers fundamental mechanisms of how cell-fate specification is interconnected to cell-cycle dynamics and provides insight into autonomous circuitries governing tissue self-formation.
Keywords: EZH2; SMYD2; TGFβ/ACTIVIN-SMAD2/3; cell cycle; differentiation; epigenetics; human pluripotent stem cells.
© The Author(s) 2024. Published by Oxford University Press on behalf of Higher Education Press.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Smad2 is essential for maintenance of the human and mouse primed pluripotent stem cell state.J Biol Chem. 2013 Jun 21;288(25):18546-60. doi: 10.1074/jbc.M112.446591. Epub 2013 May 6. J Biol Chem. 2013. PMID: 23649632 Free PMC article.
-
Derivation of trophoblast stem cells from naïve human pluripotent stem cells.Elife. 2020 Feb 12;9:e52504. doi: 10.7554/eLife.52504. Elife. 2020. PMID: 32048992 Free PMC article.
-
SMAD2 S-palmitoylation promotes its linker region phosphorylation and TH17 cell differentiation in a mouse model of multiple sclerosis.Sci Signal. 2025 May 27;18(888):eadr2008. doi: 10.1126/scisignal.adr2008. Epub 2025 May 27. Sci Signal. 2025. PMID: 40424363 Free PMC article.
-
Relationship between Pituitary Gland and Stem Cell in the Aspect of Hormone Production and Disease Prevention: A Narrative Review.Endocr Metab Immune Disord Drug Targets. 2025;25(7):509-526. doi: 10.2174/0118715303314551241031093717. Endocr Metab Immune Disord Drug Targets. 2025. PMID: 39812047 Review.
-
Management of urinary stones by experts in stone disease (ESD 2025).Arch Ital Urol Androl. 2025 Jun 30;97(2):14085. doi: 10.4081/aiua.2025.14085. Epub 2025 Jun 30. Arch Ital Urol Androl. 2025. PMID: 40583613 Review.
References
-
- Abu-Farha M, Lambert JP, Al-Madhoun AS et al. The tale of two domains: proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol Cell Proteomics 2008;7:560–572. - PubMed
-
- Awad MM, Sanders JA, Gruppuso PA. A potential role for p15(Ink4b) and p57(Kip2) in liver development. FEBS Lett 2000;483:160–164. - PubMed
-
- Blanco E, Gonzalez-Ramirez M, Alcaine-Colet A et al. The bivalent genome: characterization, structure, and regulation. Trends Genet 2020;36:118–131. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
