

PAI-1 mediates acquired resistance to MET-targeted therapy in non-small cell lung cancer
- PMID: 38758826
- PMCID: PMC11101109
- DOI: 10.1371/journal.pone.0300644
PAI-1 mediates acquired resistance to MET-targeted therapy in non-small cell lung cancer
Abstract
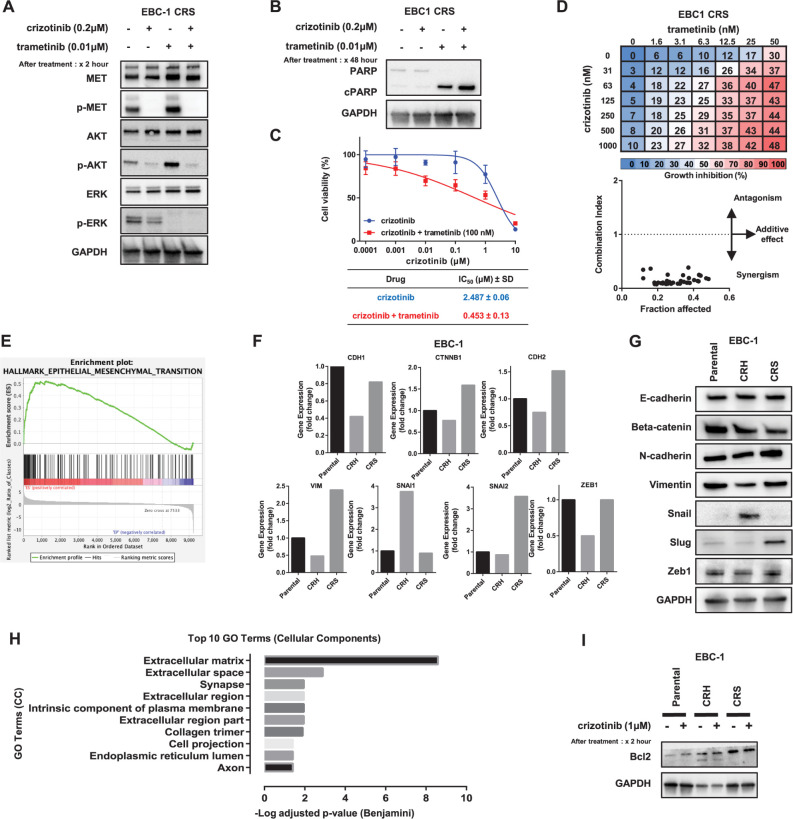
Mechanisms underlying primary and acquired resistance to MET tyrosine kinase inhibitors (TKIs) in managing non-small cell lung cancer remain unclear. In this study, we investigated the possible mechanisms acquired for crizotinib in MET-amplified lung carcinoma cell lines. Two MET-amplified lung cancer cell lines, EBC-1 and H1993, were established for acquired resistance to MET-TKI crizotinib and were functionally elucidated. Genomic and transcriptomic data were used to assess the factors contributing to the resistance mechanism, and the alterations hypothesized to confer resistance were validated. Multiple mechanisms underlie acquired resistance to crizotinib in MET-amplified lung cancer cell lines. In EBC-1-derived resistant cells, the overexpression of SERPINE1, the gene encoding plasminogen activator inhibitor-1 (PAI-1), mediated the drug resistance mechanism. Crizotinib resistance was addressed by combination therapy with a PAI-1 inhibitor and PAI-1 knockdown. Another mechanism of resistance in different subline cells of EBC-1 was evaluated as epithelial-to-mesenchymal transition with the upregulation of antiapoptotic proteins. In H1993-derived resistant cells, MEK inhibitors could be a potential therapeutic strategy for overcoming resistance with downstream mitogen-activated protein kinase pathway activation. In this study, we revealed the different mechanisms of acquired resistance to the MET inhibitor crizotinib with potential therapeutic application in patients with MET-amplified lung carcinoma.
Copyright: © 2024 Thu et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
I have read the journal’s policy and the authors of this manuscript have the following competing interests: Shinichi Toyooka received research funding from Eli Lilly Japan, Taiho (Japan) and Chugai (Japan), and lecture fees from Chugai. All other authors have declared that no competing interests exist.
Figures



Similar articles
-
Inhibition of ABCB1 Overcomes Cancer Stem Cell-like Properties and Acquired Resistance to MET Inhibitors in Non-Small Cell Lung Cancer.Mol Cancer Ther. 2015 Nov;14(11):2433-40. doi: 10.1158/1535-7163.MCT-15-0050. Epub 2015 Sep 8. Mol Cancer Ther. 2015. PMID: 26351321
-
Overcoming MET-targeted drug resistance in MET-amplified lung cancer by aurora kinase B inhibition.Biochim Biophys Acta Mol Cell Res. 2025 Oct;1872(7):120001. doi: 10.1016/j.bbamcr.2025.120001. Epub 2025 Jun 9. Biochim Biophys Acta Mol Cell Res. 2025. PMID: 40499687
-
Acquired Resistance of MET-Amplified Non-small Cell Lung Cancer Cells to the MET Inhibitor Capmatinib.Cancer Res Treat. 2019 Jul;51(3):951-962. doi: 10.4143/crt.2018.052. Epub 2018 Oct 10. Cancer Res Treat. 2019. PMID: 30309221 Free PMC article.
-
Targeting the MET gene for the treatment of non-small-cell lung cancer.Crit Rev Oncol Hematol. 2014 Feb;89(2):284-99. doi: 10.1016/j.critrevonc.2013.11.006. Epub 2013 Dec 1. Crit Rev Oncol Hematol. 2014. PMID: 24355409 Review.
-
Current mechanism of acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and updated therapy strategies in human nonsmall cell lung cancer.J Cancer Res Ther. 2016 Dec;12(Supplement):C131-C137. doi: 10.4103/0973-1482.200613. J Cancer Res Ther. 2016. PMID: 28230005 Review.
Cited by
-
Unconventional localization of PAI-1 in PML bodies: A possible link with cellular growth of endothelial cells.Biochem Biophys Rep. 2024 Jul 26;39:101793. doi: 10.1016/j.bbrep.2024.101793. eCollection 2024 Sep. Biochem Biophys Rep. 2024. PMID: 39161580 Free PMC article.
-
Phosphorylation of MET Is Upregulated in Metastatic Sites of Renal Cell Carcinoma: Possible Role of MET and Hepatocyte Growth Factor Activation-Targeted Combined Therapy.Biomedicines. 2025 Mar 28;13(4):811. doi: 10.3390/biomedicines13040811. Biomedicines. 2025. PMID: 40299443 Free PMC article.
-
MiR-145-5p inhibits proliferation of hepatocellular carcinoma, acting through PAI-1.Am J Transl Res. 2025 Feb 15;17(2):888-896. doi: 10.62347/NPLT8946. eCollection 2025. Am J Transl Res. 2025. PMID: 40092137 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
