

Genome-wide DNA methylation changes in human spermatogenesis
- PMID: 38759652
- PMCID: PMC11179423
- DOI: 10.1016/j.ajhg.2024.04.017
Genome-wide DNA methylation changes in human spermatogenesis
Abstract
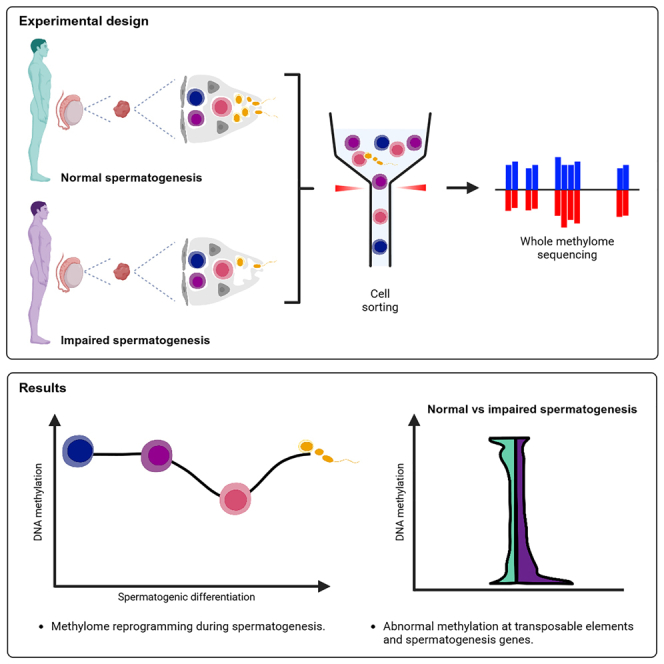
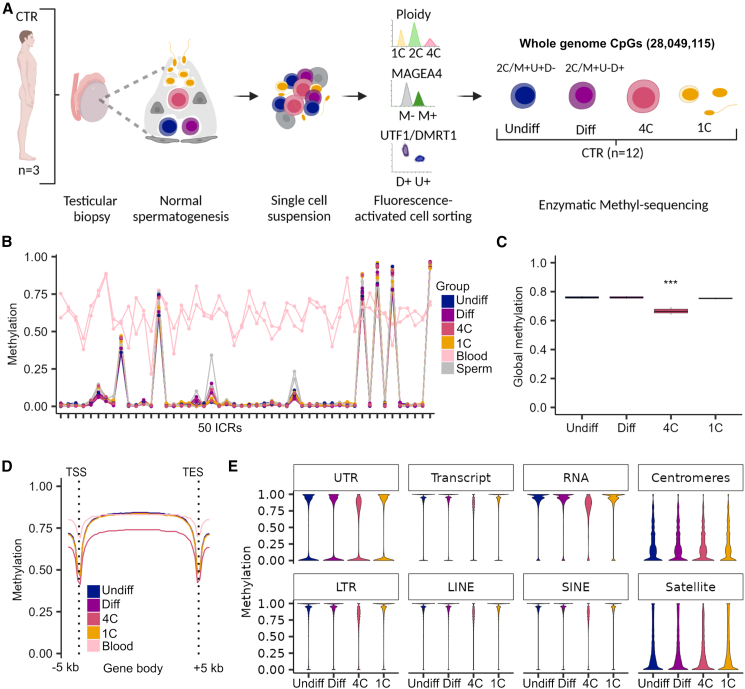
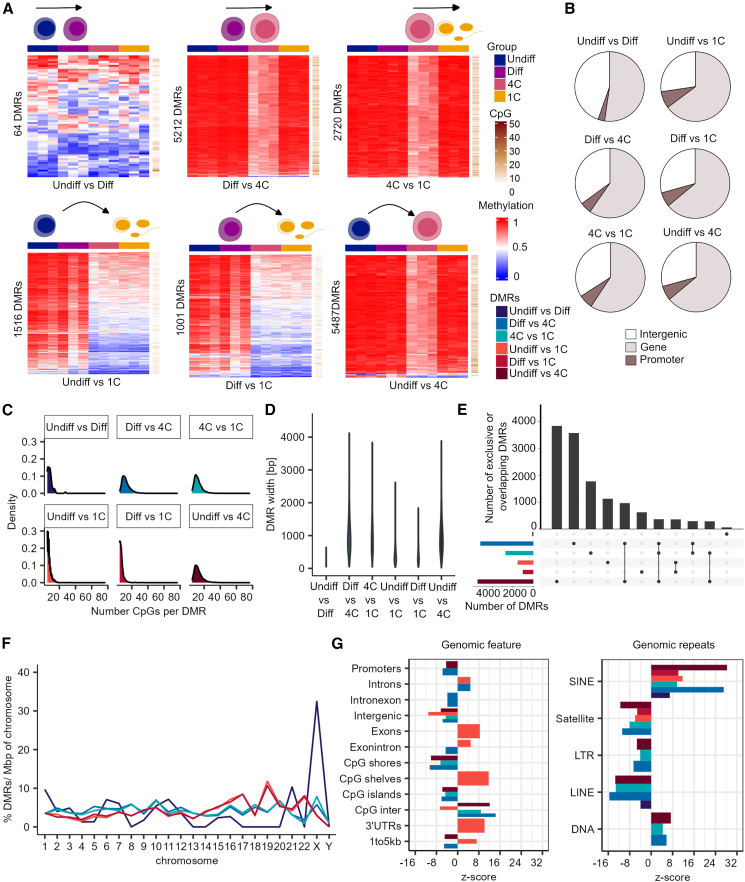
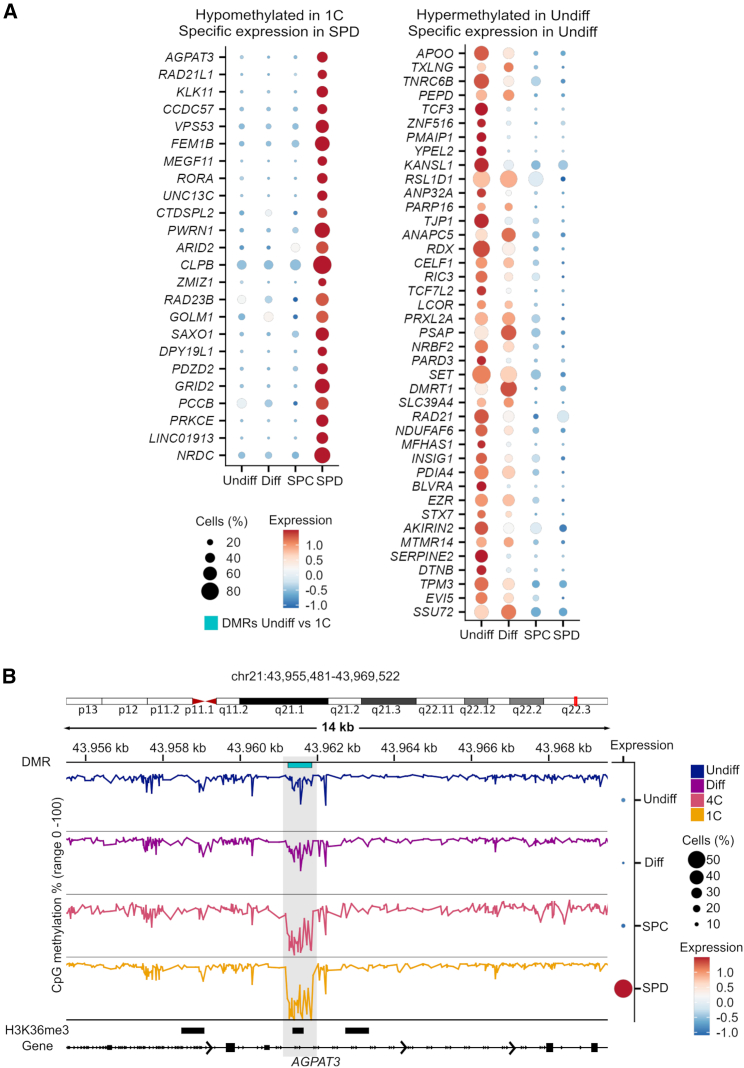
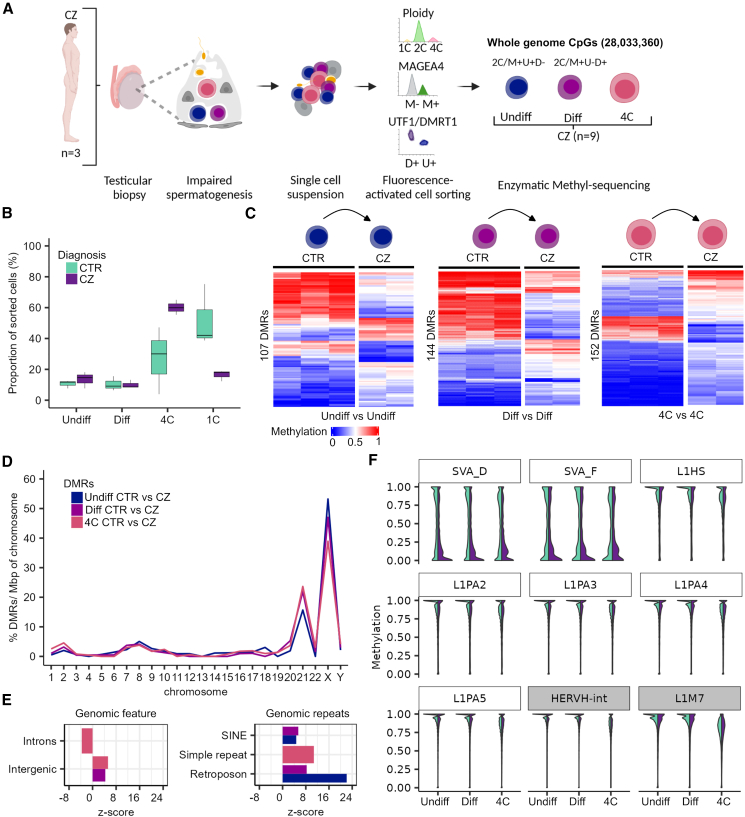
Sperm production and function require the correct establishment of DNA methylation patterns in the germline. Here, we examined the genome-wide DNA methylation changes during human spermatogenesis and its alterations in disturbed spermatogenesis. We found that spermatogenesis is associated with remodeling of the methylome, comprising a global decline in DNA methylation in primary spermatocytes followed by selective remethylation, resulting in a spermatids/sperm-specific methylome. Hypomethylated regions in spermatids/sperm were enriched in specific transcription factor binding sites for DMRT and SOX family members and spermatid-specific genes. Intriguingly, while SINEs displayed differential methylation throughout spermatogenesis, LINEs appeared to be protected from changes in DNA methylation. In disturbed spermatogenesis, germ cells exhibited considerable DNA methylation changes, which were significantly enriched at transposable elements and genes involved in spermatogenesis. We detected hypomethylation in SVA and L1HS in disturbed spermatogenesis, suggesting an association between the abnormal programming of these regions and failure of germ cells progressing beyond meiosis.
Keywords: DNA methylation; epigenetics; germline; human spermatogenesis; infertility; male germ cells; methylome; transposable elements.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures

References
-
- Langenstroth-Röwer D., Gromoll J., Wistuba J., Tröndle I., Laurentino S., Schlatt S., Neuhaus N. De novo methylation in male germ cells of the common marmoset monkey occurs during postnatal development and is maintained in vitro. Epigenetics. 2017;12:527–539. doi: 10.1080/15592294.2016.1248007. - DOI - PMC - PubMed
-
- Di Persio S., Leitão E., Wöste M., Tekath T., Cremers J.-F., Dugas M., Li X., Meyer zu Hörste G., Kliesch S., Laurentino S., et al. Whole-genome methylation analysis of testicular germ cells from cryptozoospermic men points to recurrent and functionally relevant DNA methylation changes. Clin. Epigenet. 2021;13:160. doi: 10.1186/s13148-021-01144-z. - DOI - PMC - PubMed
-
- Huang Y., Li L., An G., Yang X., Cui M., Song X., Lin J., Zhang X., Yao Z., Wan C., et al. Single-cell multi-omics sequencing of human spermatogenesis reveals a DNA demethylation event associated with male meiotic recombination. Nat. Cell Biol. 2023;25:1520–1534. doi: 10.1038/s41556-023-01232-7. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
