

Deficiency of ADAR2 ameliorates metabolic-associated fatty liver disease via AMPK signaling pathways in obese mice
- PMID: 38760406
- PMCID: PMC11101631
- DOI: 10.1038/s42003-024-06215-4
Deficiency of ADAR2 ameliorates metabolic-associated fatty liver disease via AMPK signaling pathways in obese mice
Abstract
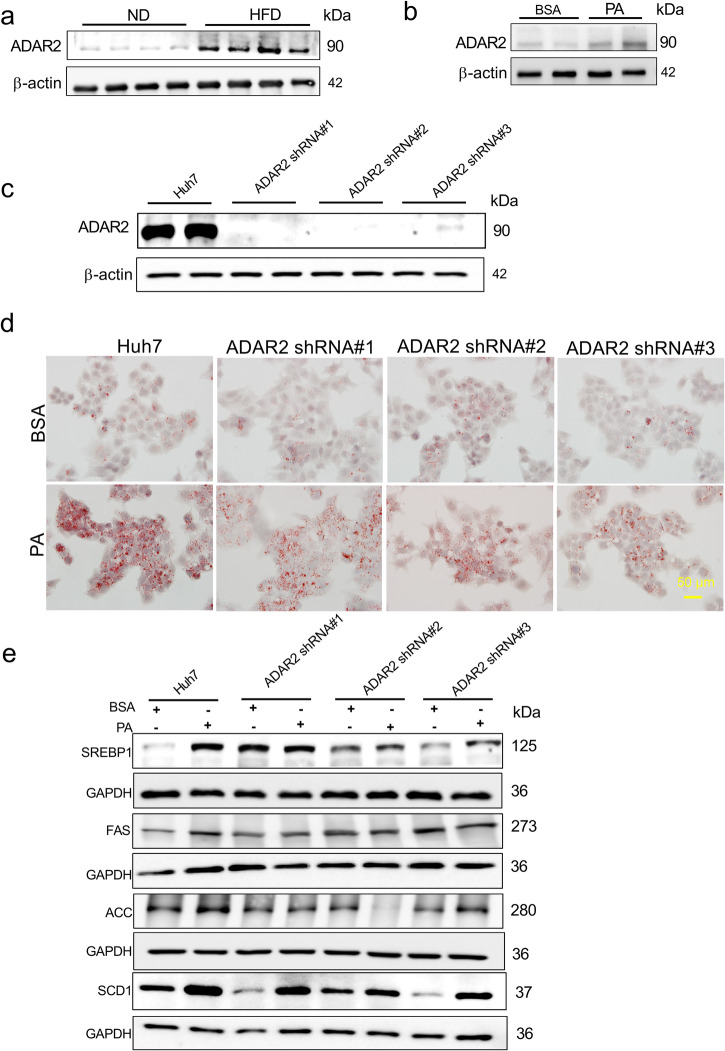
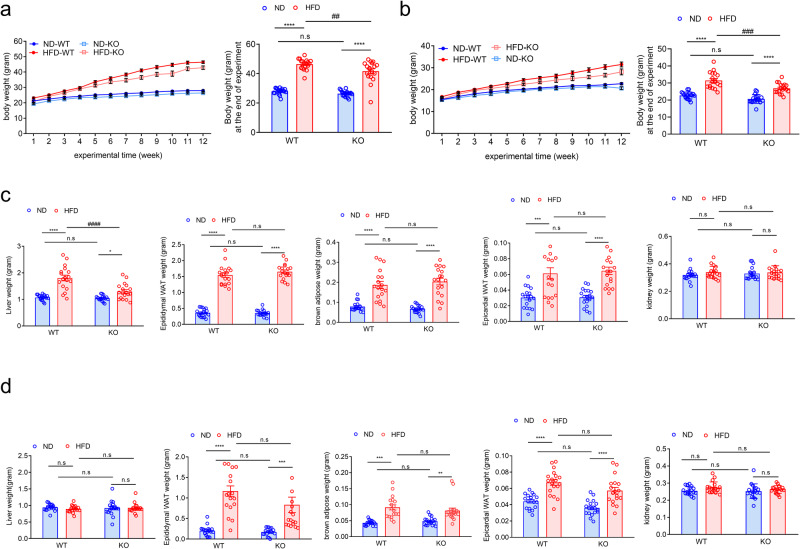
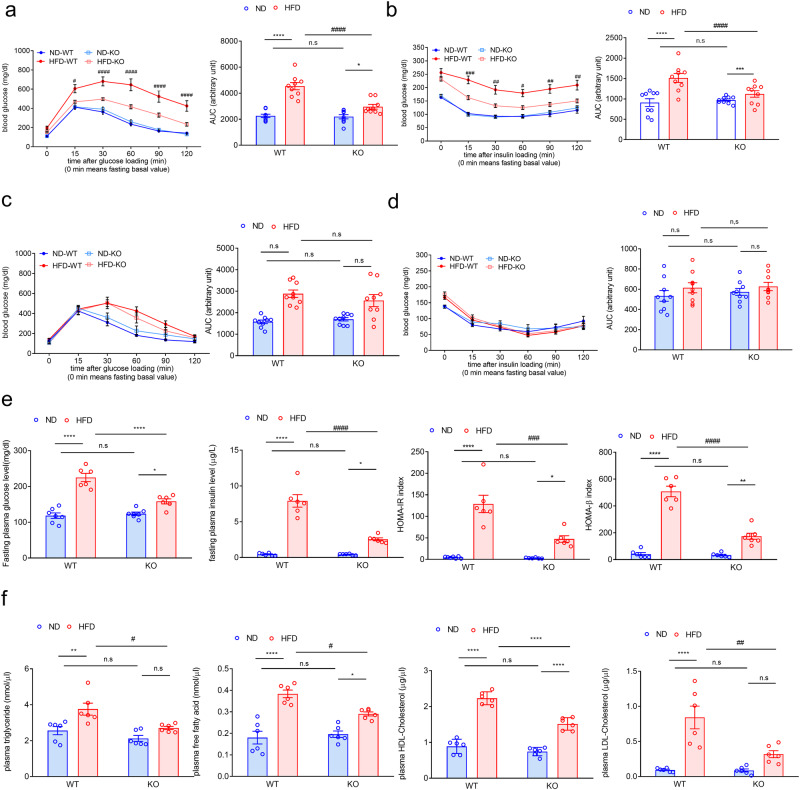
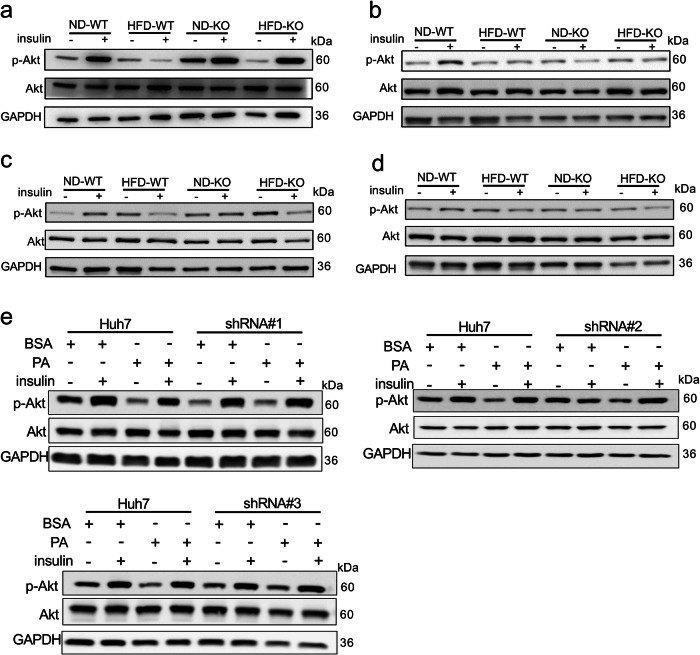
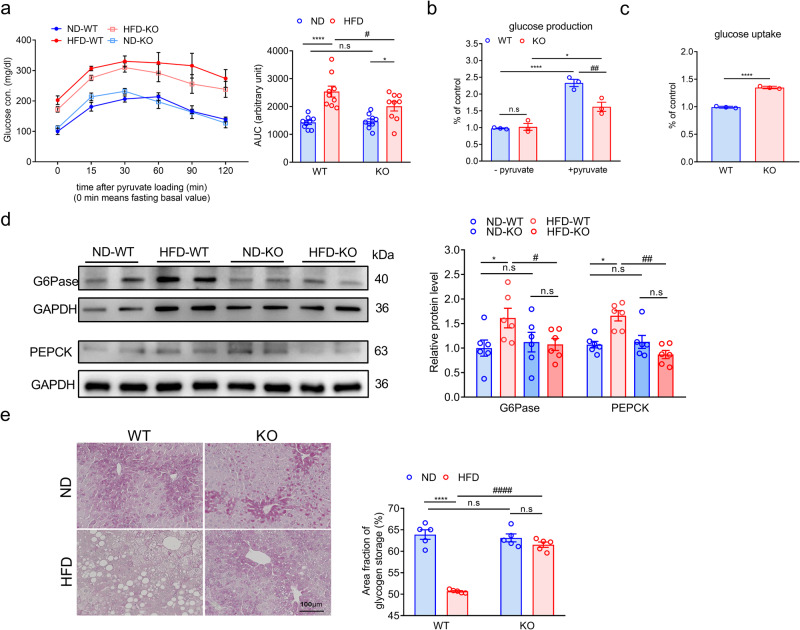
Non-alcoholic fatty liver disease (NAFLD) is a chronic disease caused by hepatic steatosis. Adenosine deaminases acting on RNA (ADARs) catalyze adenosine to inosine RNA editing. However, the functional role of ADAR2 in NAFLD is unclear. ADAR2+/+/GluR-BR/R mice (wild type, WT) and ADAR2-/-/GluR-BR/R mice (ADAR2 KO) mice are fed with standard chow or high-fat diet (HFD) for 12 weeks. ADAR2 KO mice exhibit protection against HFD-induced glucose intolerance, insulin resistance, and dyslipidemia. Moreover, ADAR2 KO mice display reduced liver lipid droplets in concert with decreased hepatic TG content, improved hepatic insulin signaling, better pyruvate tolerance, and increased glycogen synthesis. Mechanistically, ADAR2 KO effectively mitigates excessive lipid production via AMPK/Sirt1 pathway. ADAR2 KO inhibits hepatic gluconeogenesis via the AMPK/CREB pathway and promotes glycogen synthesis by activating the AMPK/GSK3β pathway. These results provide evidence that ADAR2 KO protects against NAFLD progression through the activation of AMPK signaling pathways.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
- MOST 109-2320-B-006-041-MY3/Ministry of Science and Technology, Taiwan (Ministry of Science and Technology of Taiwan)
- MOST 106-2320-B-006-058-MY3/Ministry of Science and Technology, Taiwan (Ministry of Science and Technology of Taiwan)
- MOST 112-2320-B-006-029-MY3/Ministry of Science and Technology, Taiwan (Ministry of Science and Technology of Taiwan)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
