

Spatial genomic, biochemical and cellular mechanisms underlying meningioma heterogeneity and evolution
- PMID: 38760638
- PMCID: PMC11239374
- DOI: 10.1038/s41588-024-01747-1
Spatial genomic, biochemical and cellular mechanisms underlying meningioma heterogeneity and evolution
Abstract
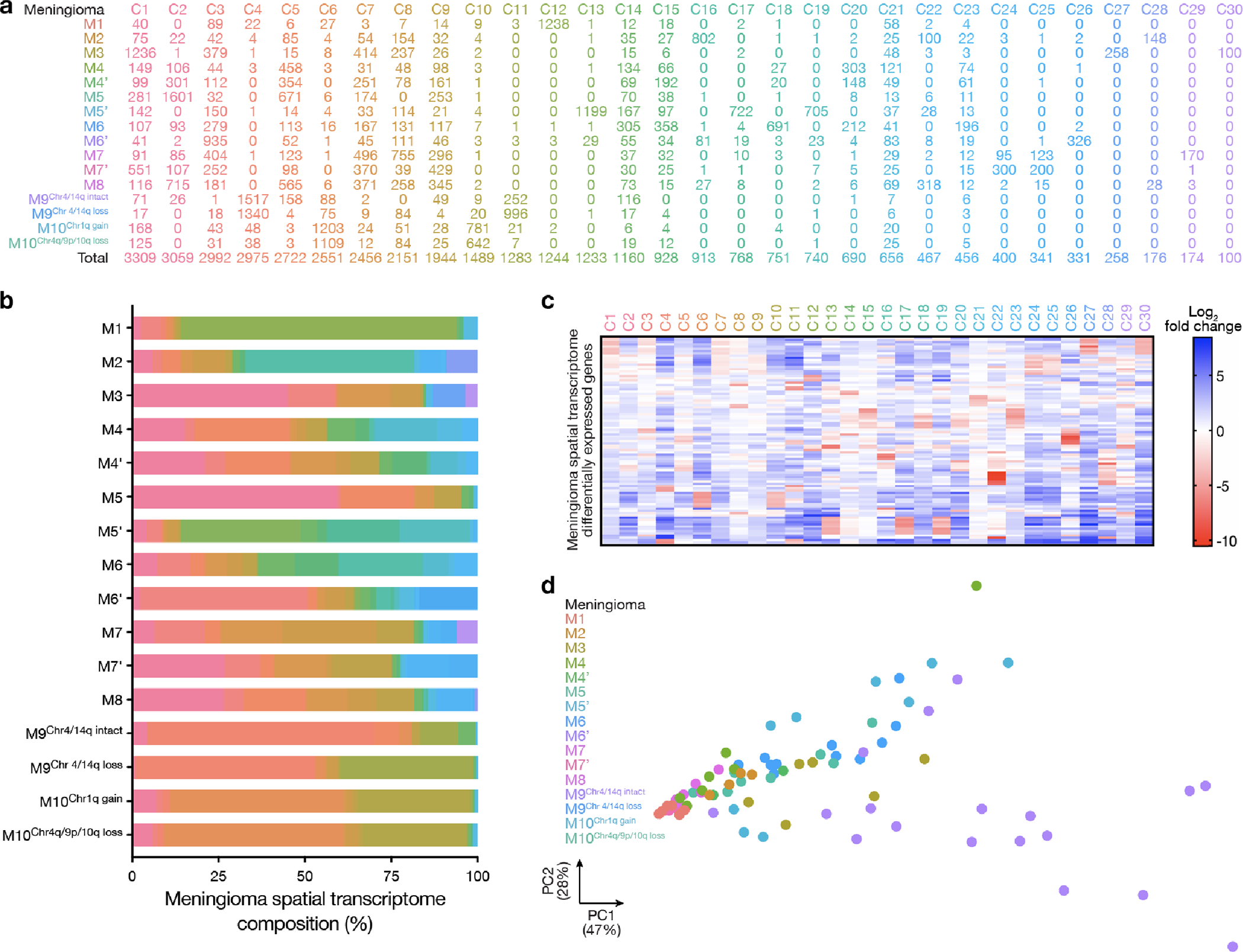
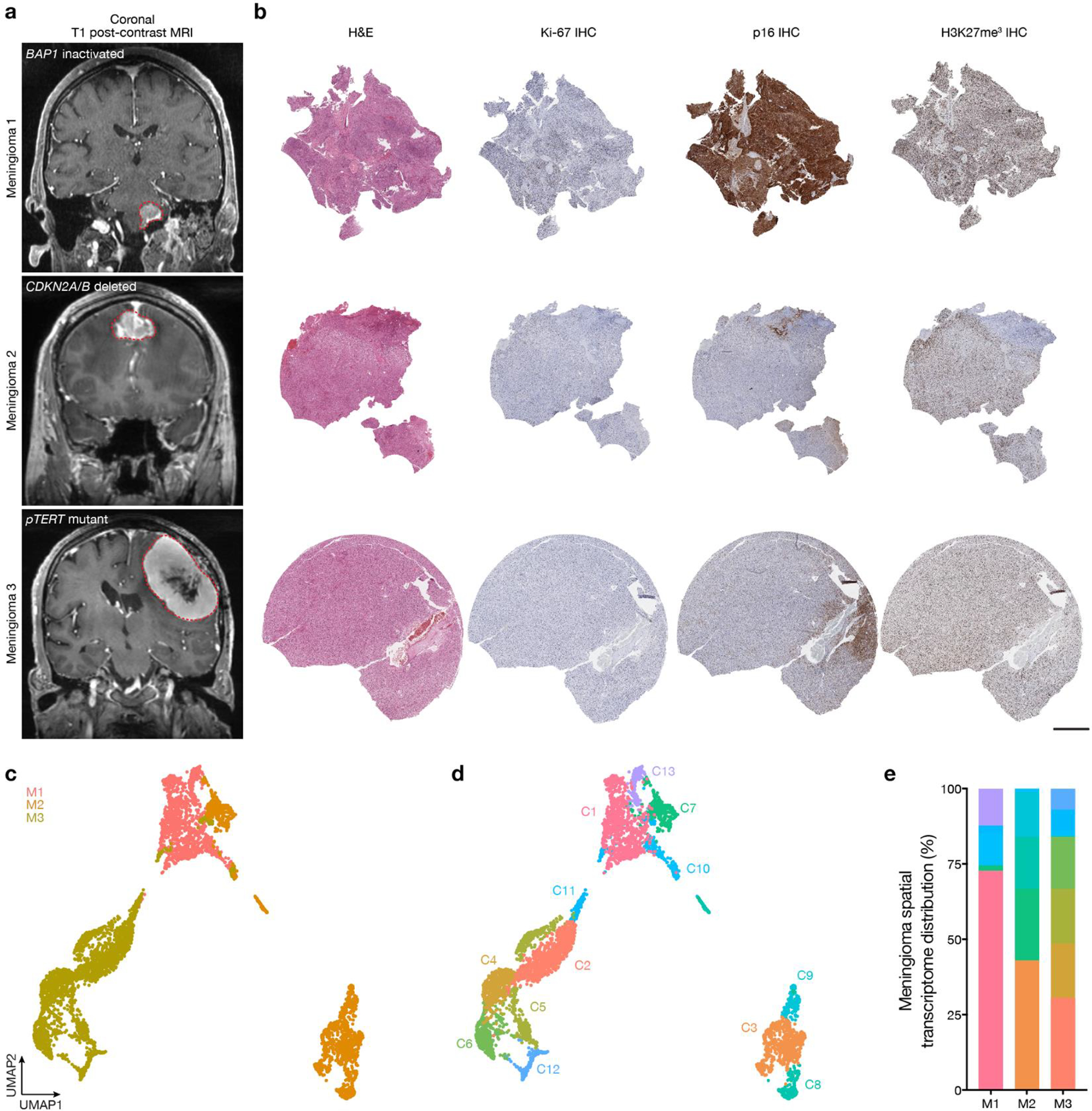
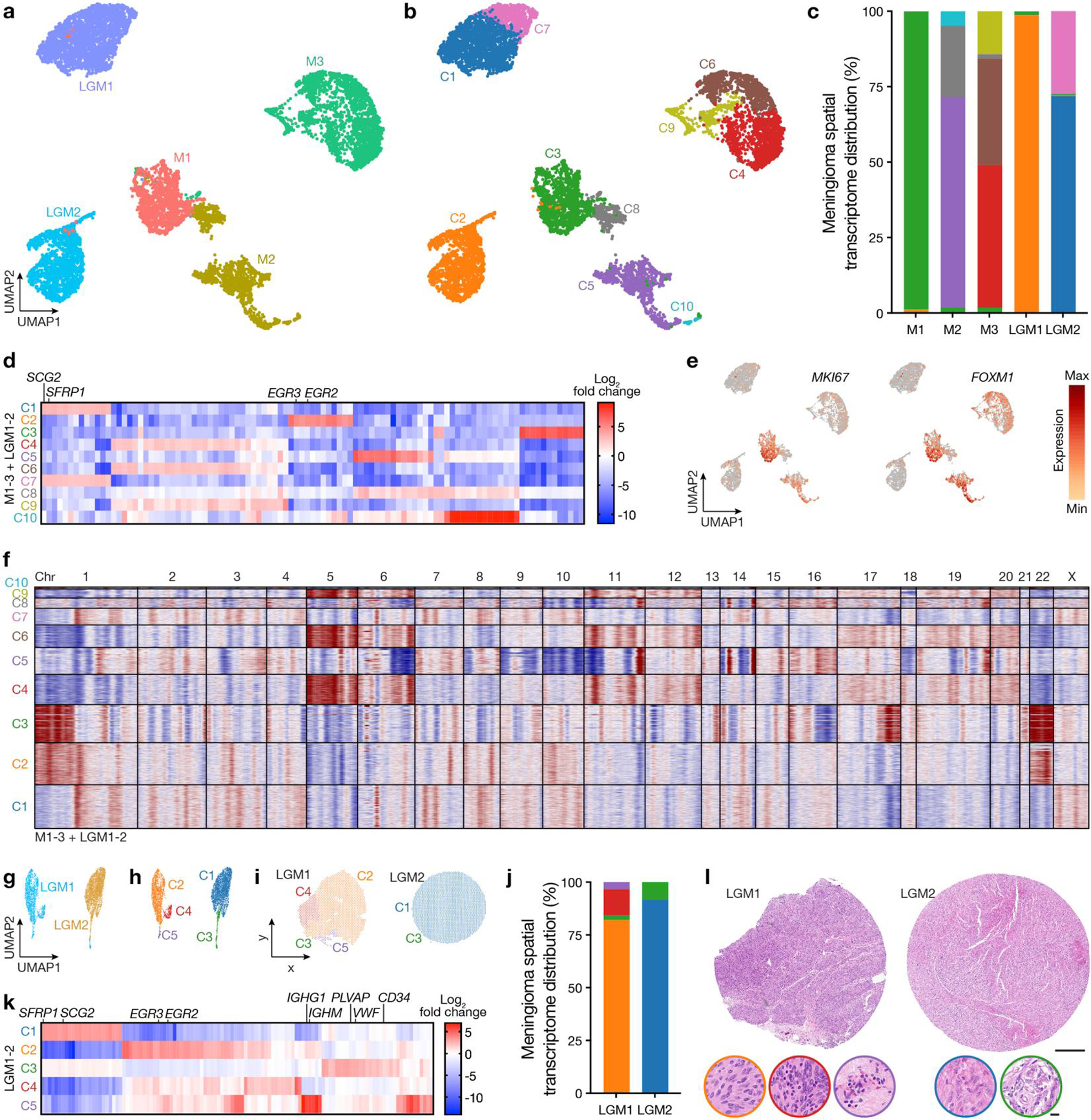
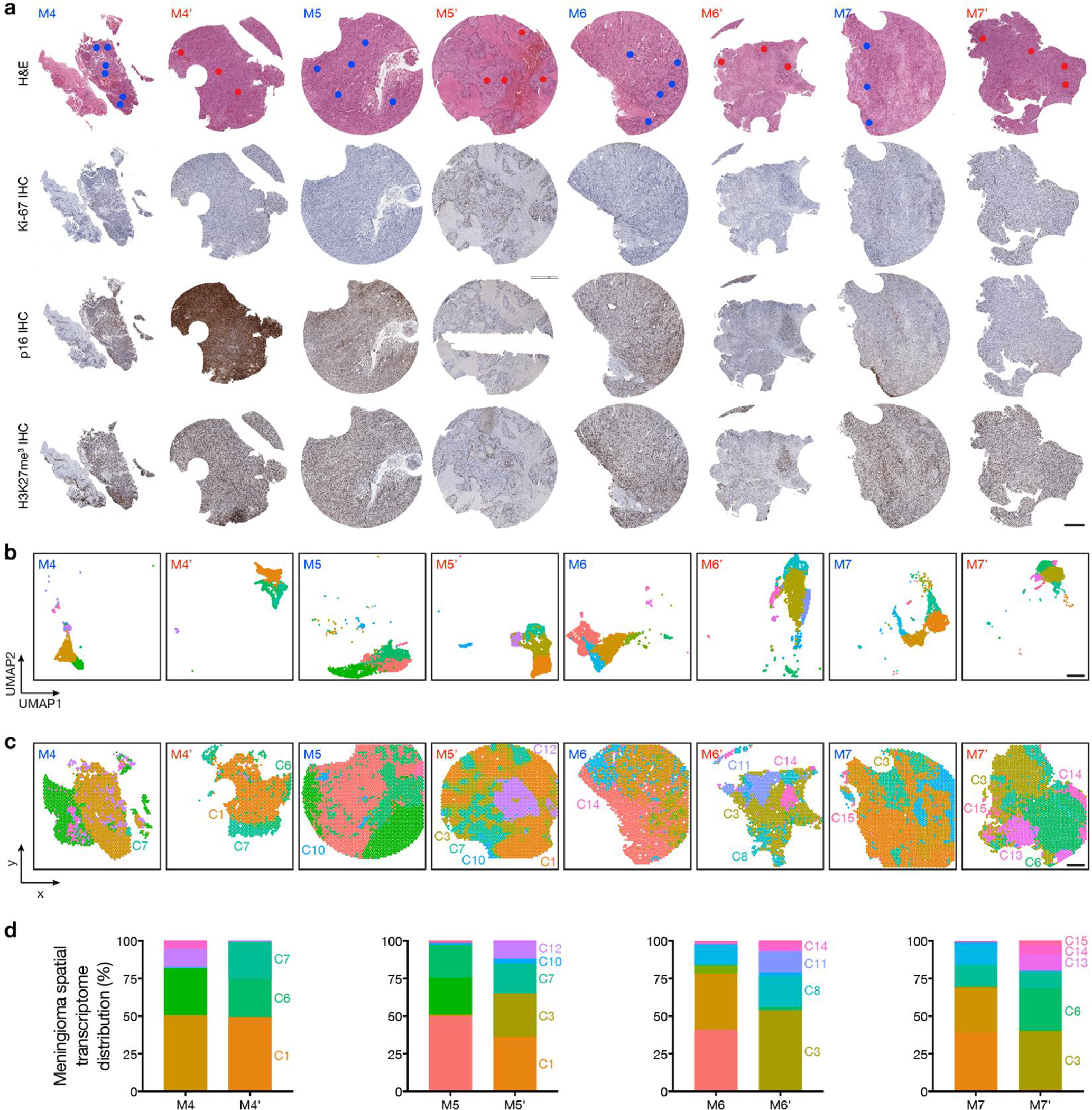
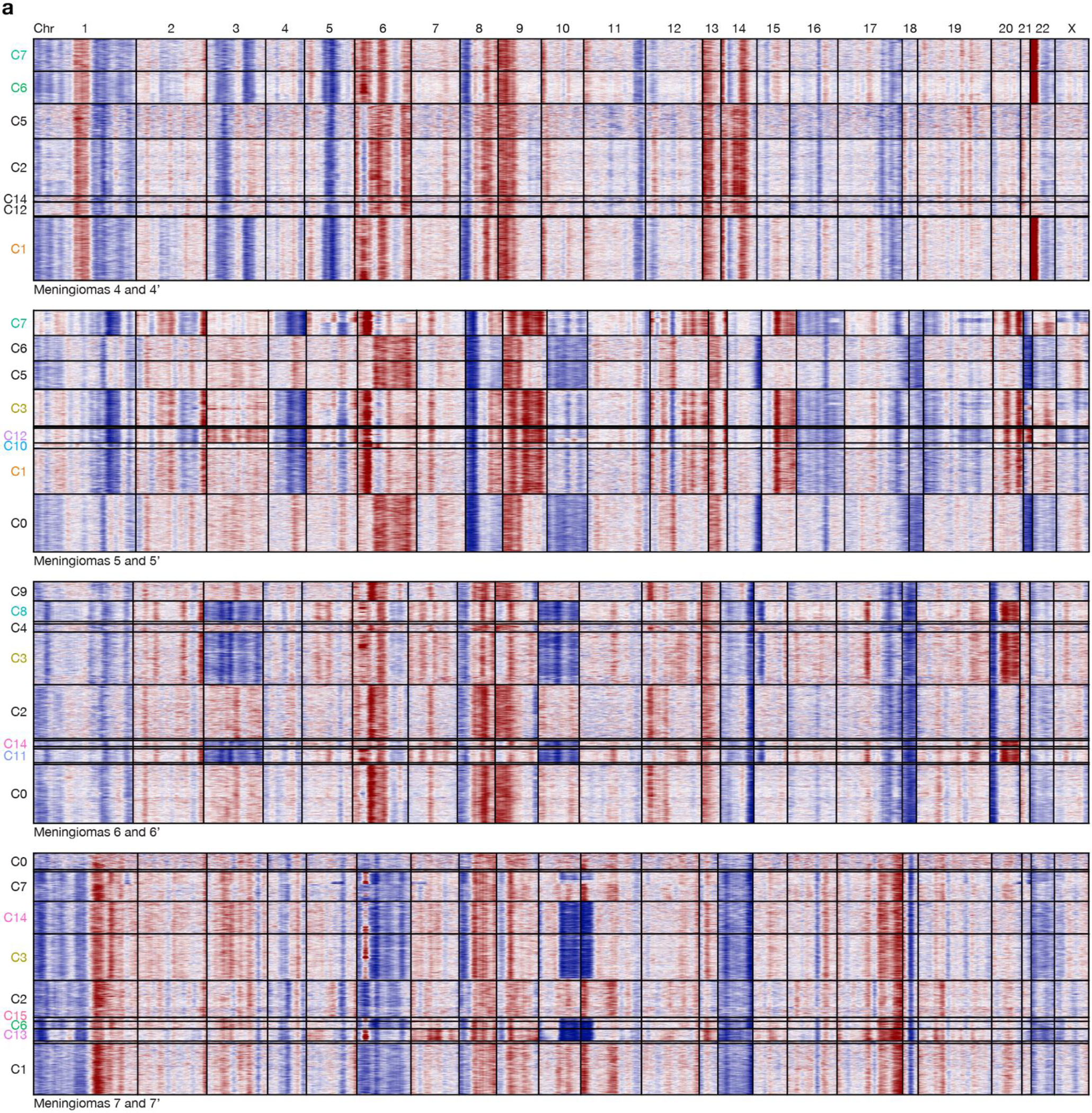
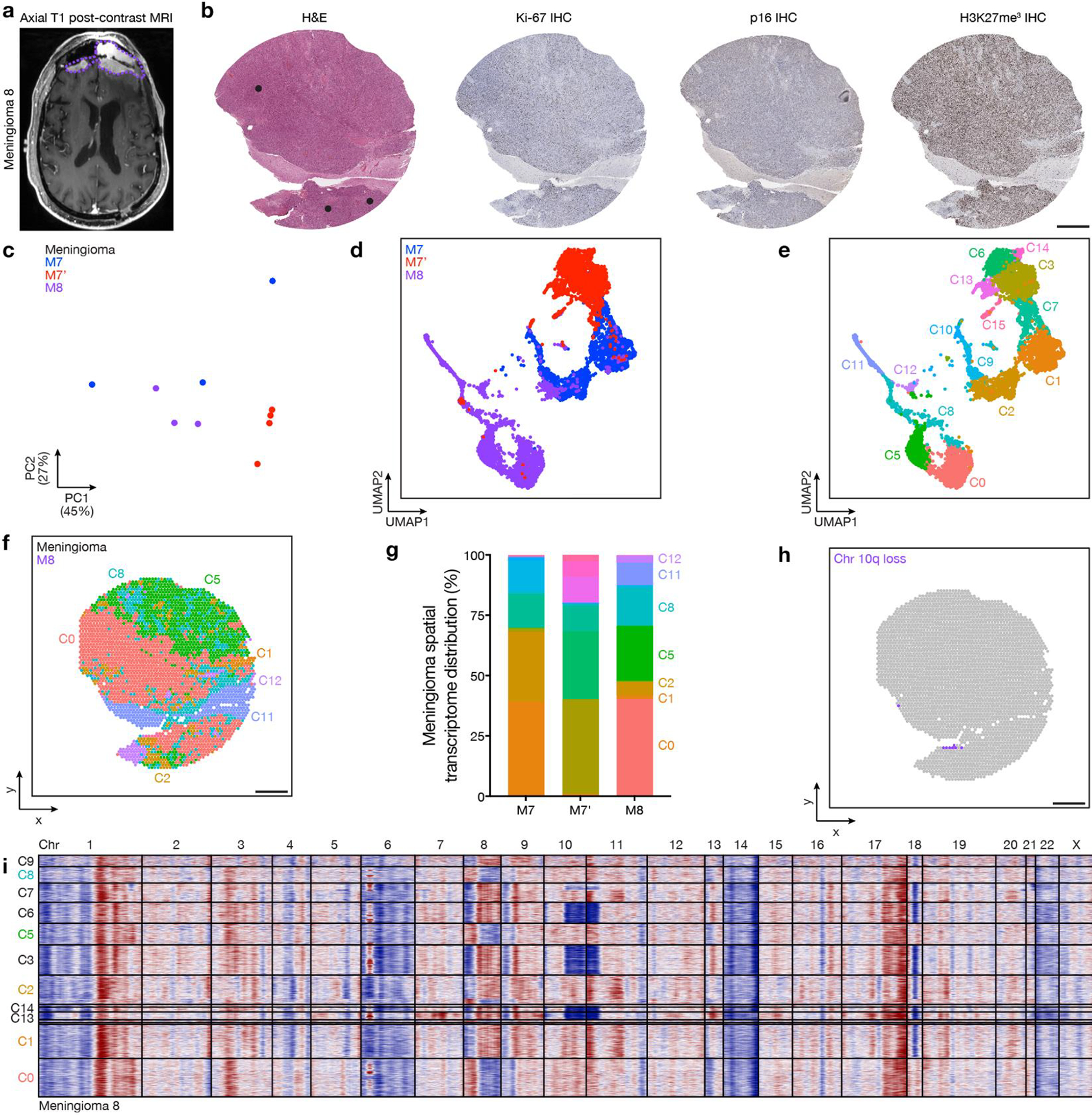
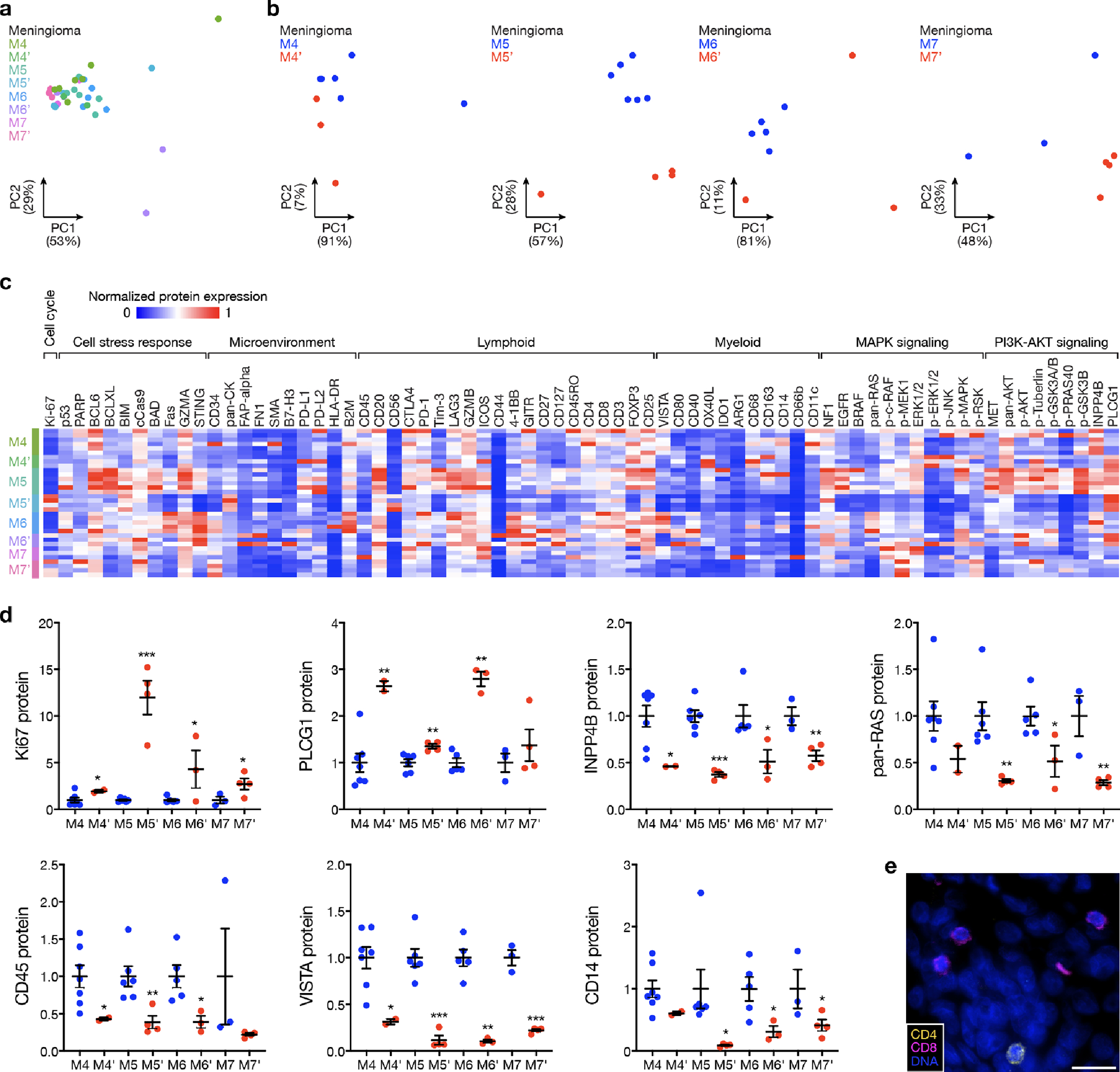
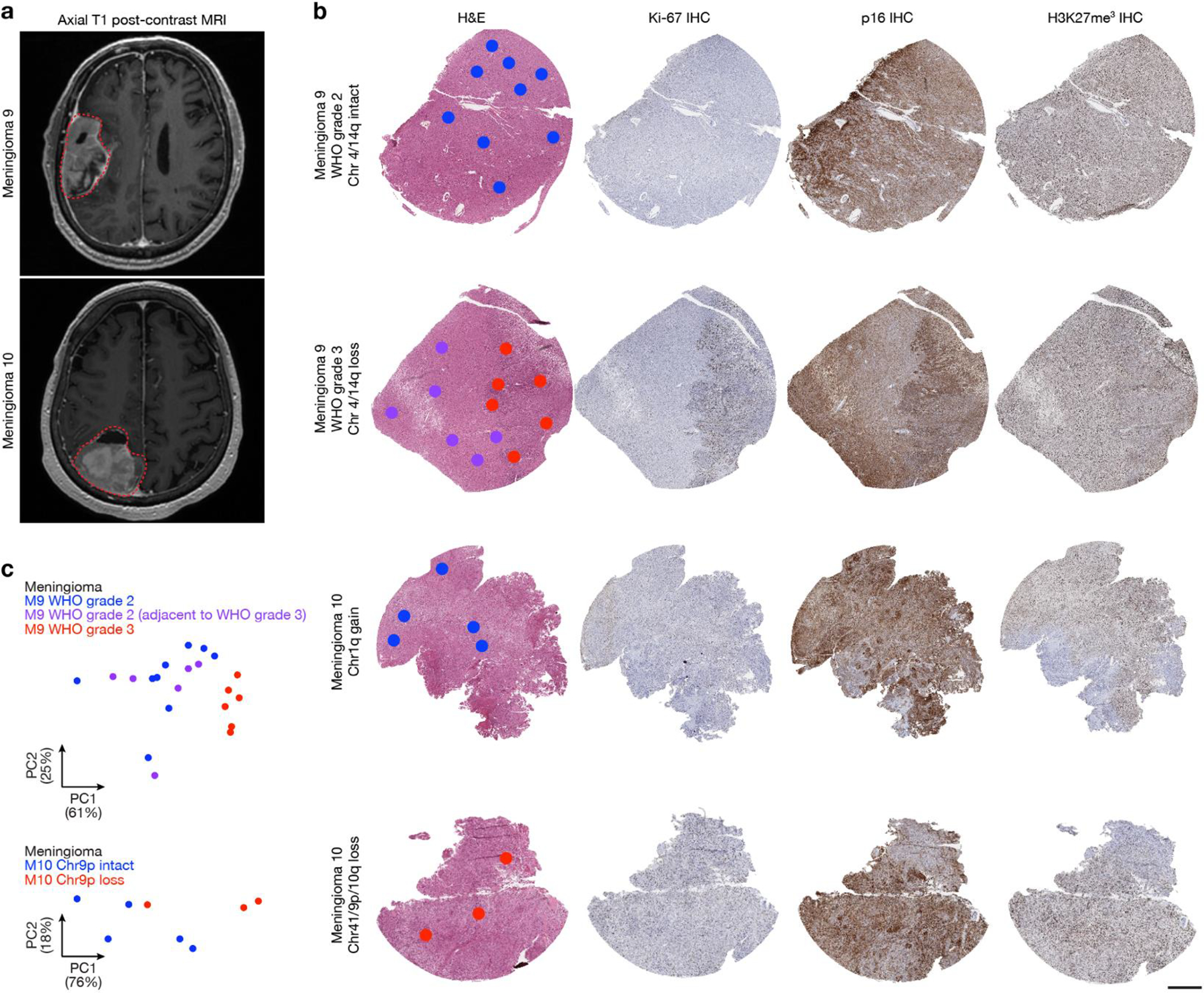
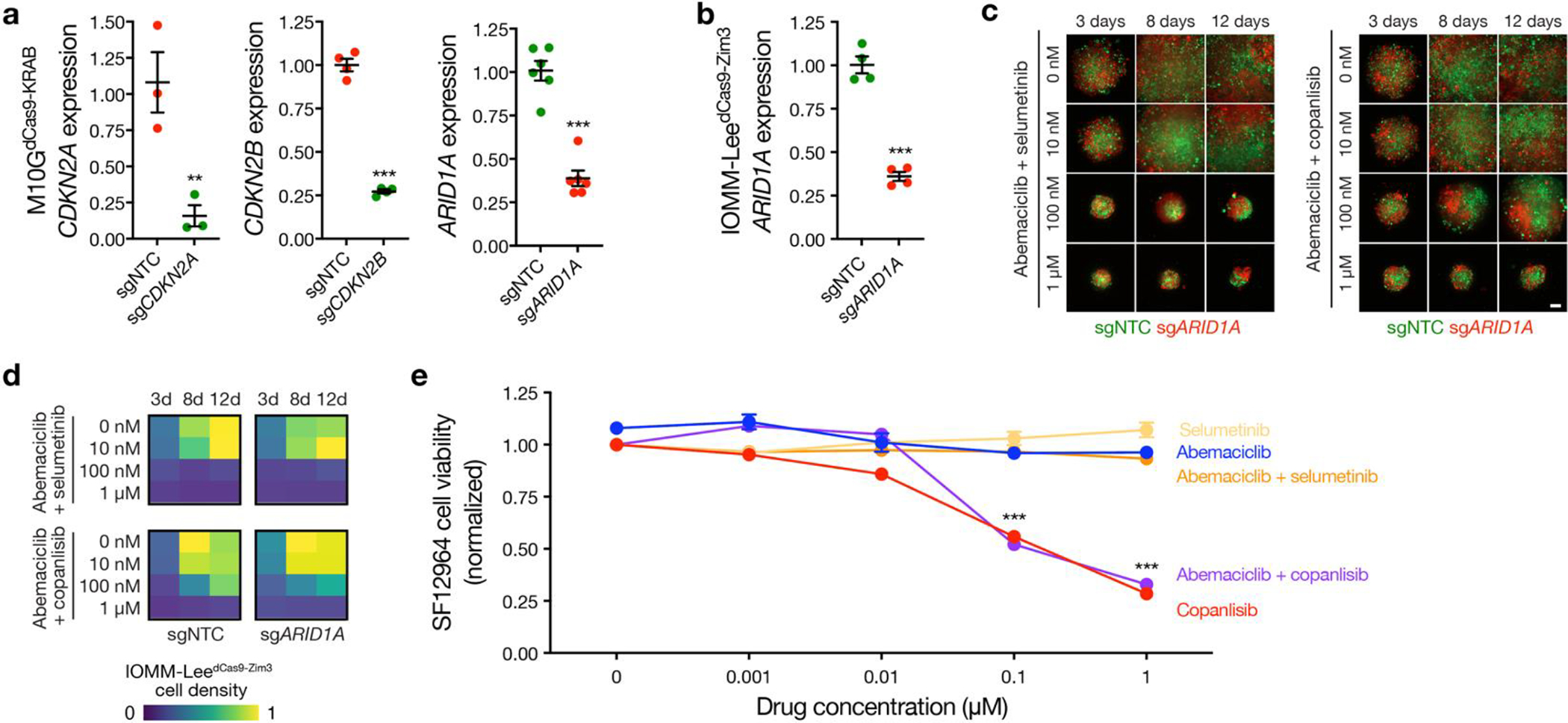
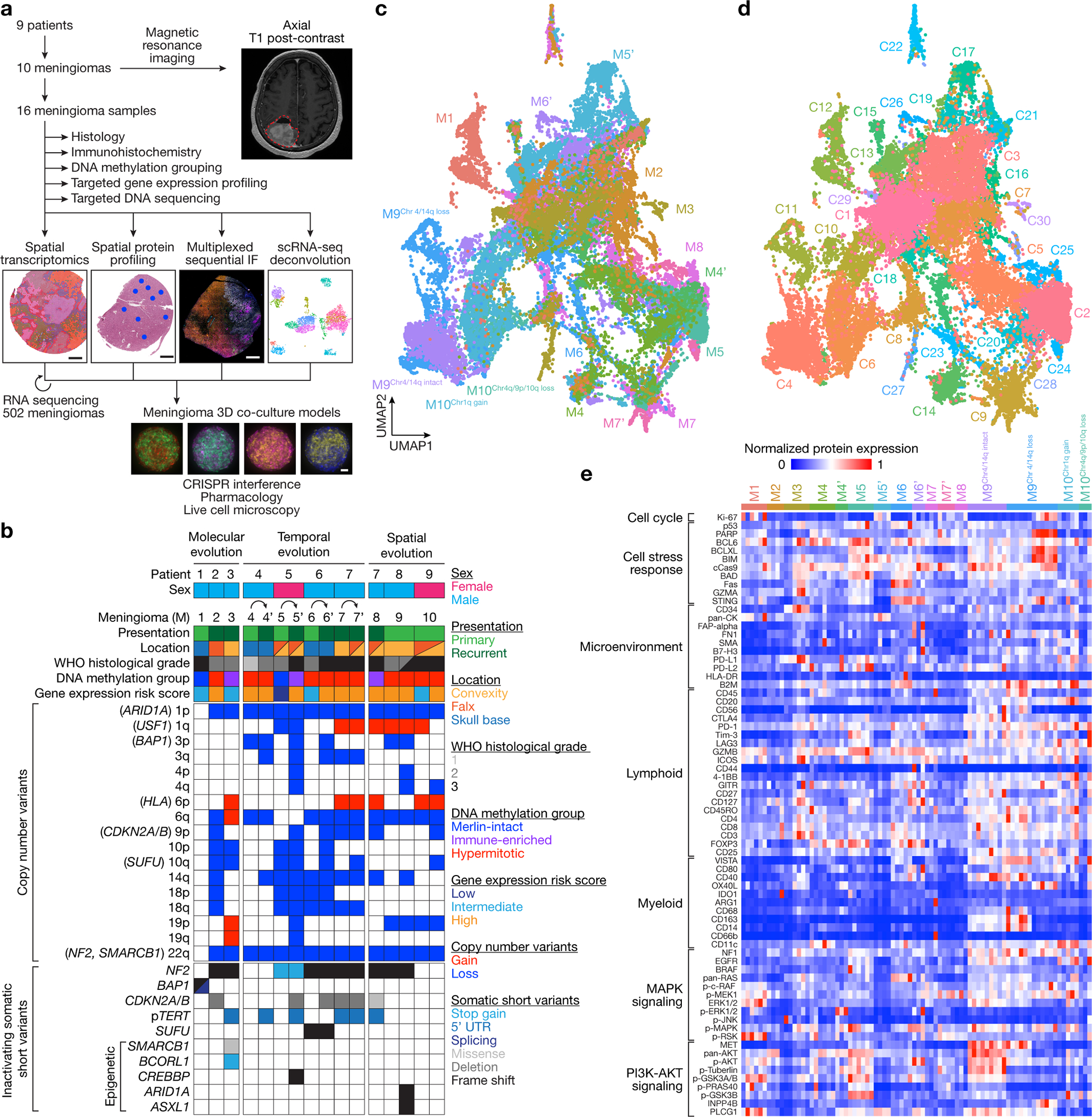
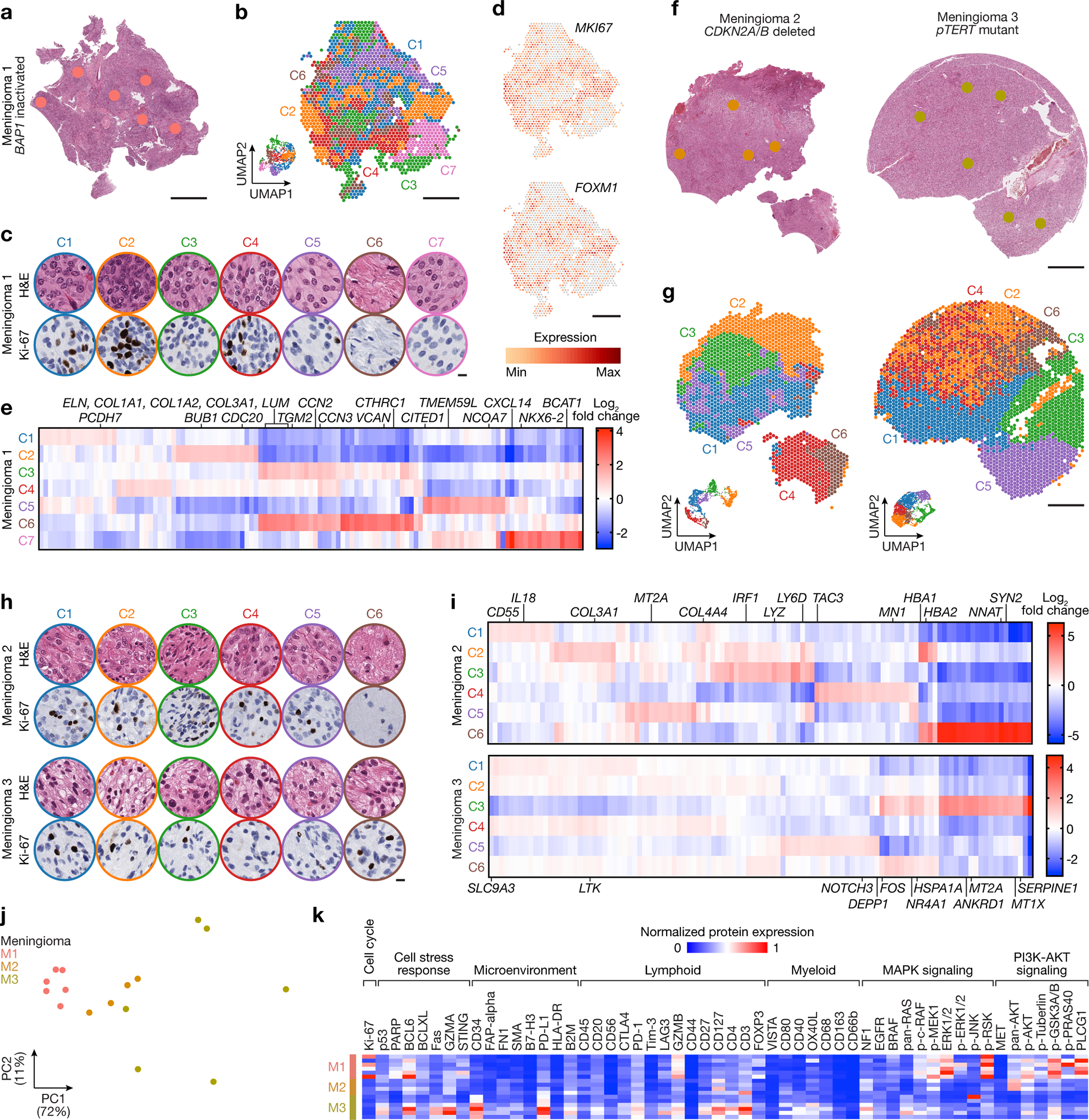
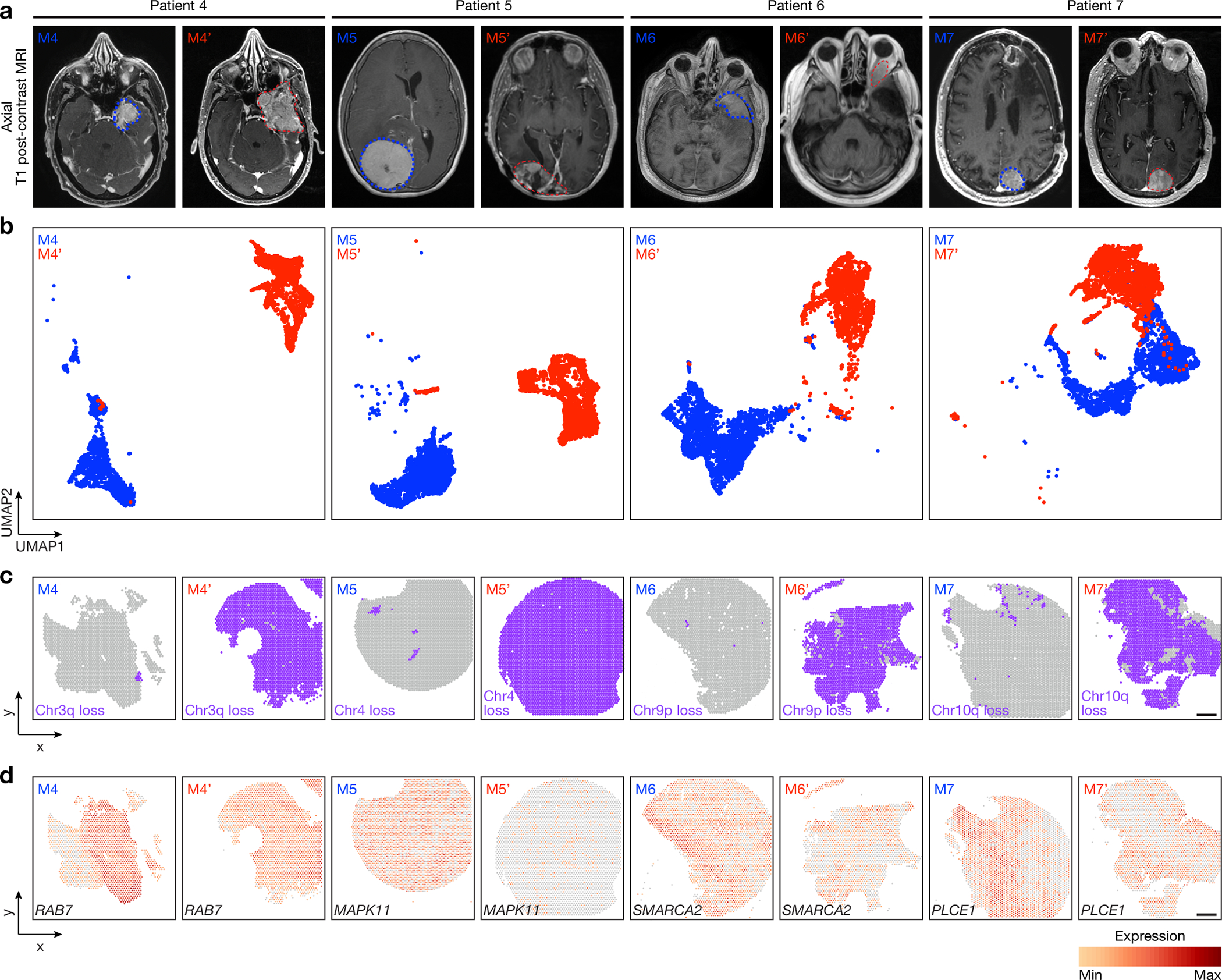
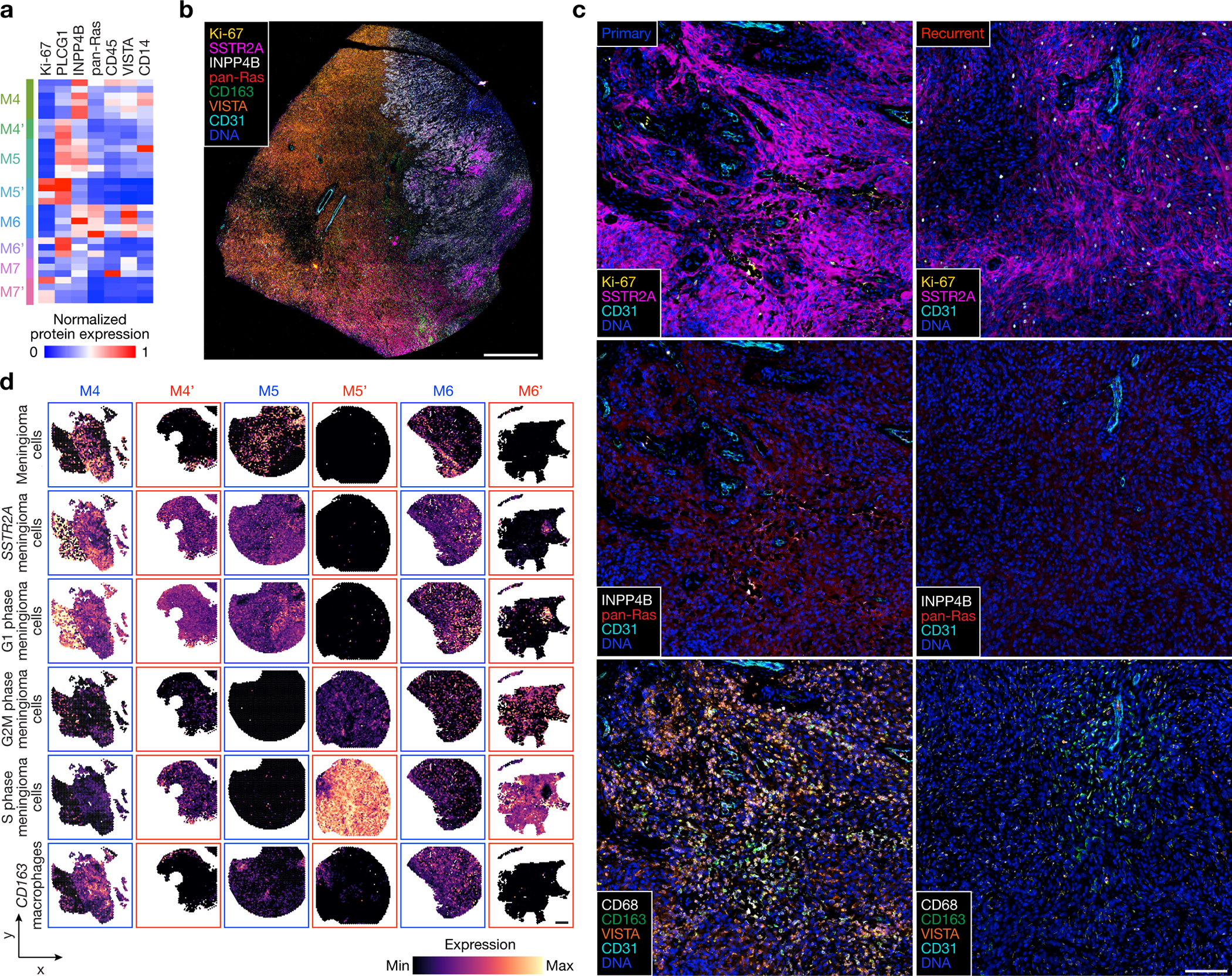
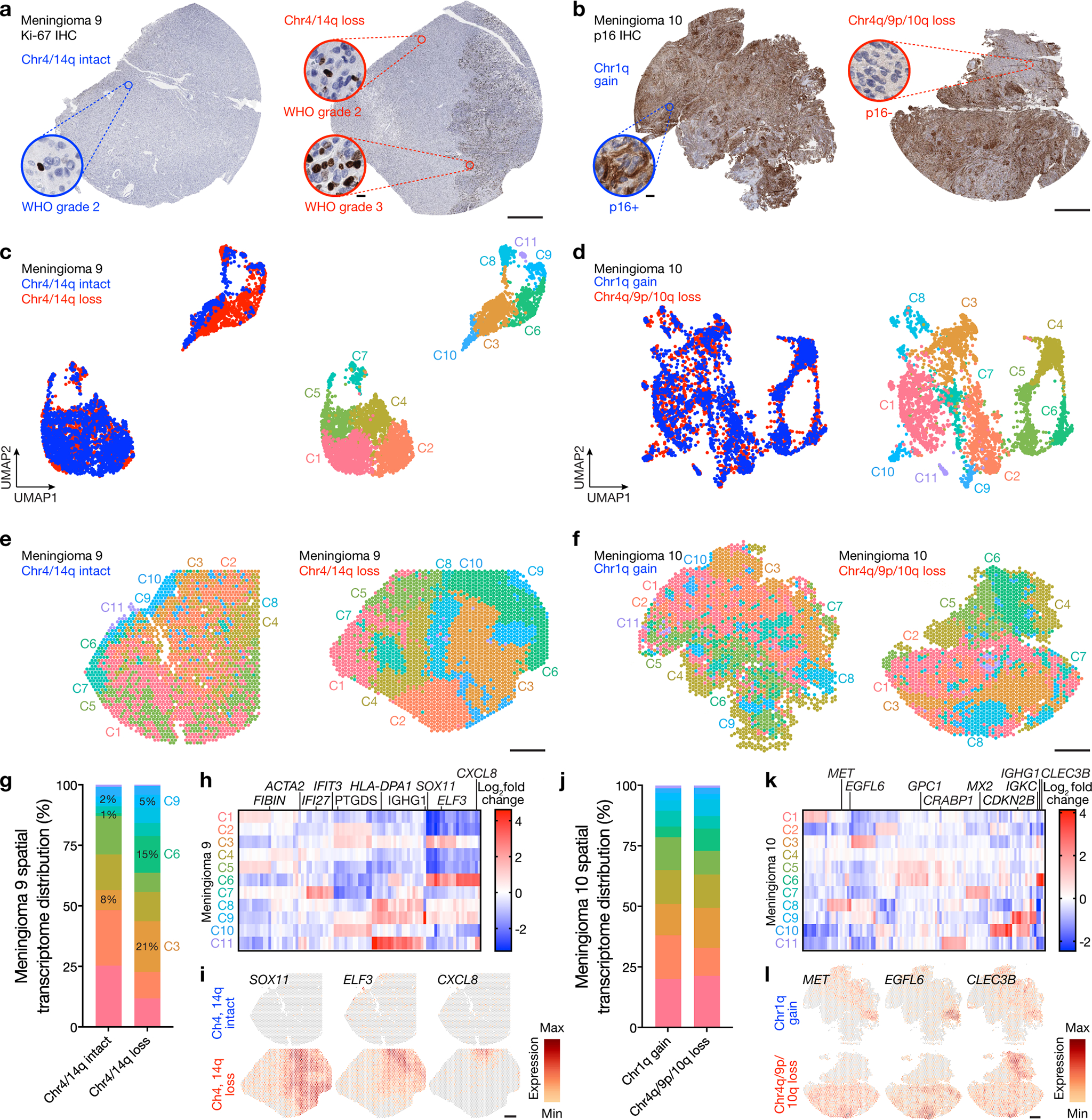
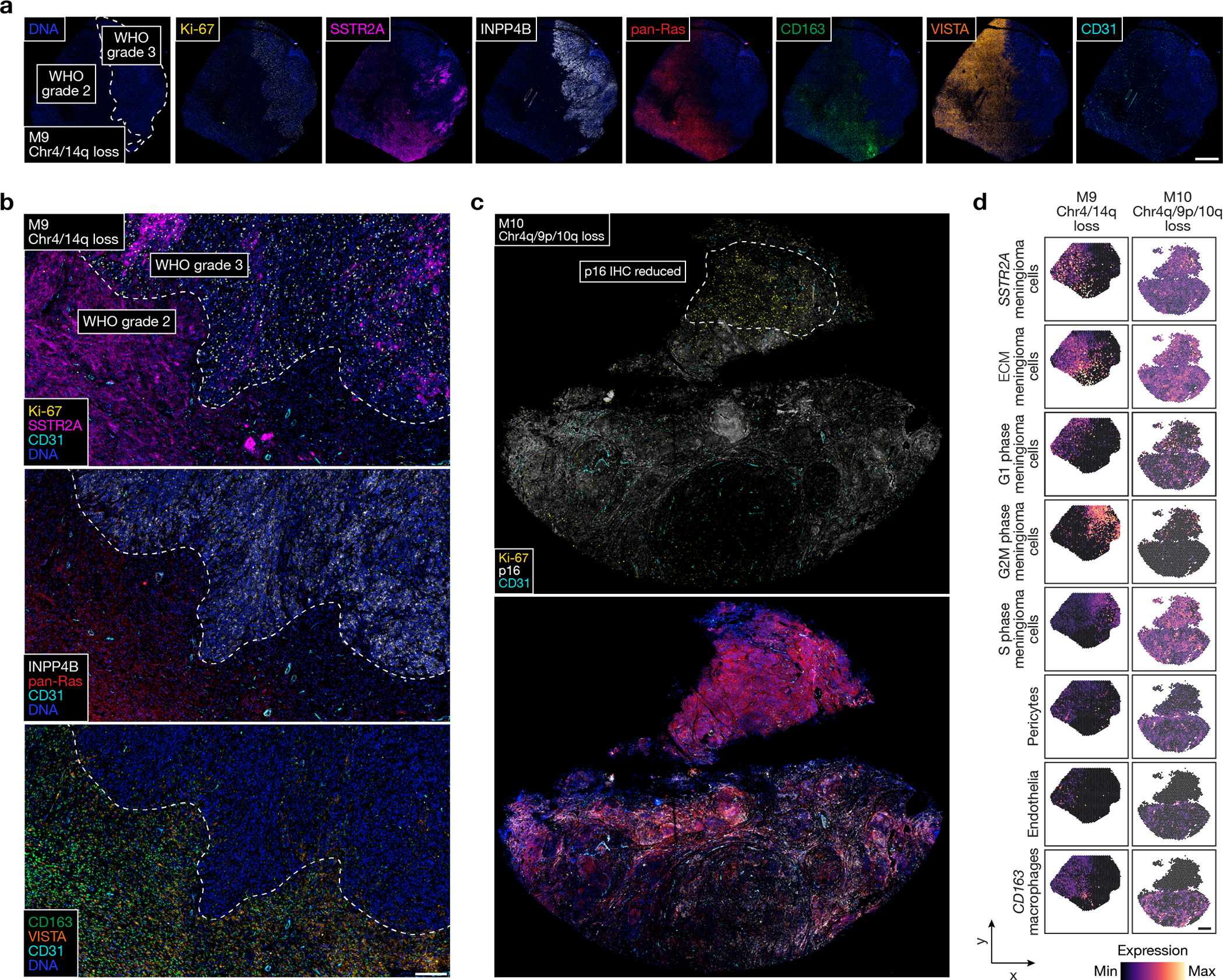
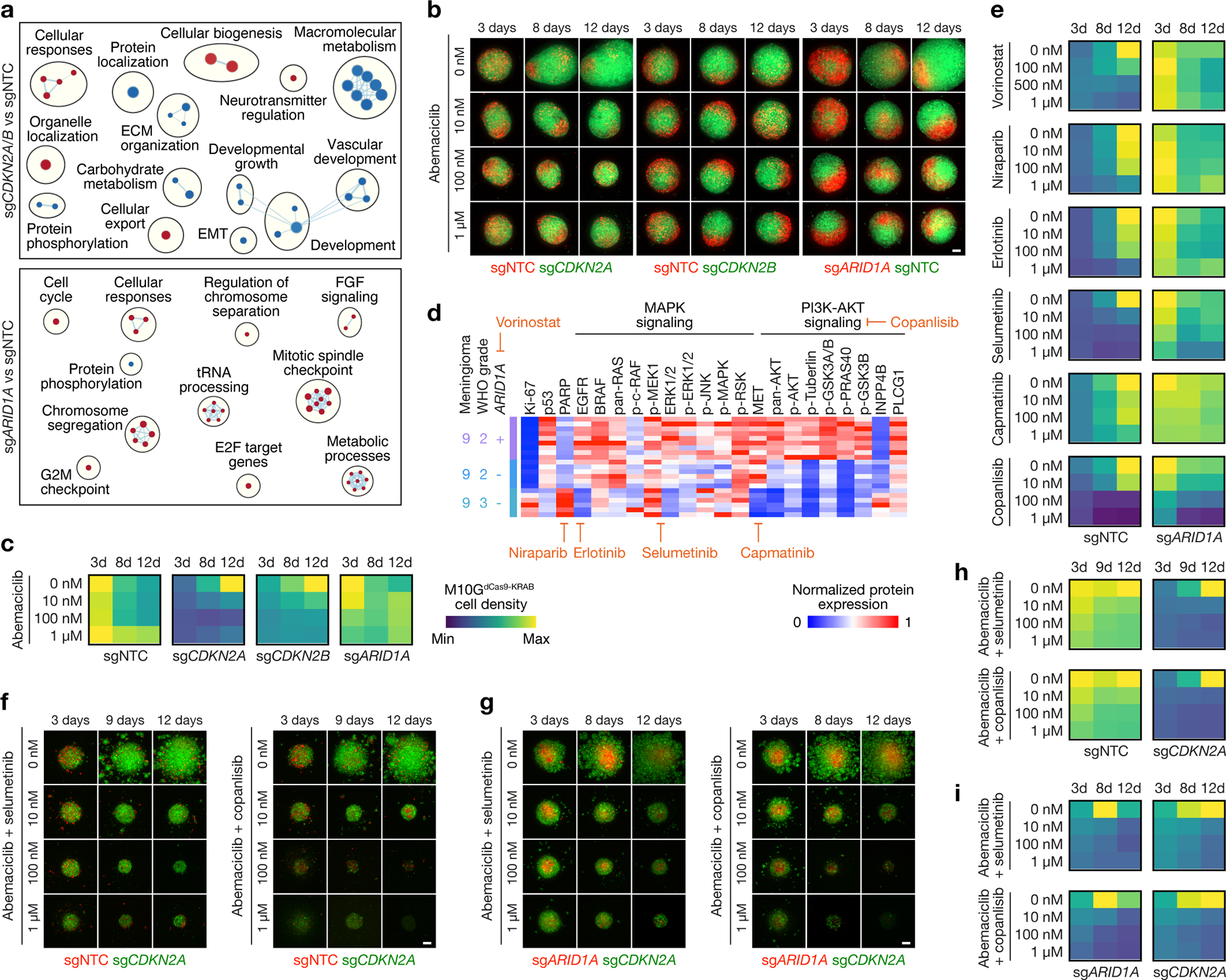
Intratumor heterogeneity underlies cancer evolution and treatment resistance, but targetable mechanisms driving intratumor heterogeneity are poorly understood. Meningiomas are the most common primary intracranial tumors and are resistant to all medical therapies, and high-grade meningiomas have significant intratumor heterogeneity. Here we use spatial approaches to identify genomic, biochemical and cellular mechanisms linking intratumor heterogeneity to the molecular, temporal and spatial evolution of high-grade meningiomas. We show that divergent intratumor gene and protein expression programs distinguish high-grade meningiomas that are otherwise grouped together by current classification systems. Analyses of matched pairs of primary and recurrent meningiomas reveal spatial expansion of subclonal copy number variants associated with treatment resistance. Multiplexed sequential immunofluorescence and deconvolution of meningioma spatial transcriptomes using cell types from single-cell RNA sequencing show decreased immune infiltration, decreased MAPK signaling, increased PI3K-AKT signaling and increased cell proliferation, which are associated with meningioma recurrence. To translate these findings to preclinical models, we use CRISPR interference and lineage tracing approaches to identify combination therapies that target intratumor heterogeneity in meningioma cell co-cultures.
© 2024. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests statement
The authors declare no competing interests.
Figures

Update of
-
Spatial genomic, biochemical, and cellular mechanisms drive meningioma heterogeneity and evolution.Res Sq [Preprint]. 2023 May 15:rs.3.rs-2921804. doi: 10.21203/rs.3.rs-2921804/v1. Res Sq. 2023. Update in: Nat Genet. 2024 Jun;56(6):1121-1133. doi: 10.1038/s41588-024-01747-1. PMID: 37292686 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
