

Persistent gene expression and DNA methylation alterations linked to carcinogenic effects of dichloroacetic acid
- PMID: 38764585
- PMCID: PMC11099211
- DOI: 10.3389/fonc.2024.1389634
Persistent gene expression and DNA methylation alterations linked to carcinogenic effects of dichloroacetic acid
Abstract
Background: Mechanistic understanding of transient exposures that lead to adverse health outcomes will enhance our ability to recognize biological signatures of disease. Here, we measured the transcriptomic and epigenomic alterations due to exposure to the metabolic reprogramming agent, dichloroacetic acid (DCA). Previously, we showed that exposure to DCA increased liver tumor incidence in B6C3F1 mice after continuous or early life exposures significantly over background level.
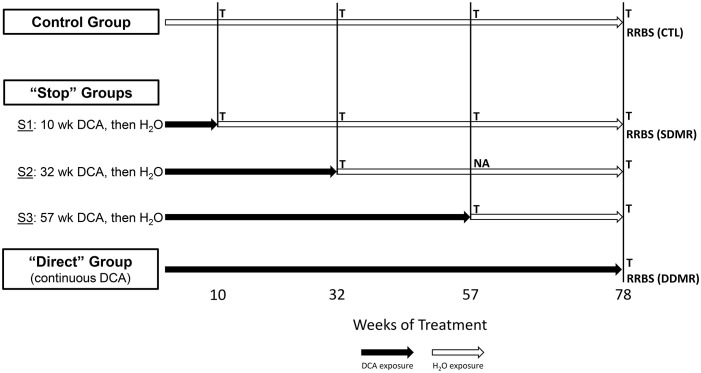
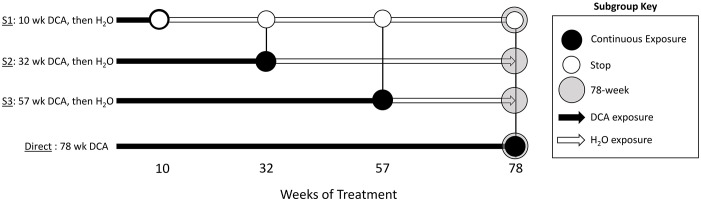
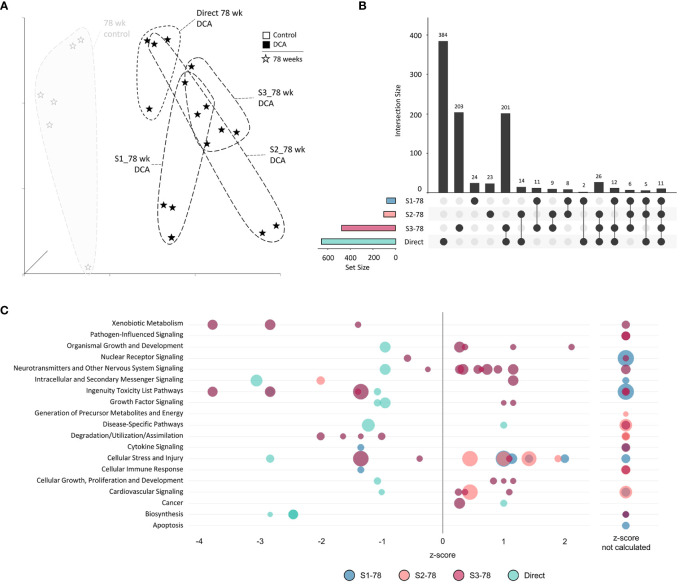
Methods: Using archived formalin-fixed liver samples, we utilized modern methodologies to measure gene expression and DNA methylation levels to link to previously generated phenotypic measures. Gene expression was measured by targeted RNA sequencing (TempO-seq 1500+ toxicity panel: 2754 total genes) in liver samples collected from 10-, 32-, 57-, and 78-week old mice exposed to deionized water (controls), 3.5 g/L DCA continuously in drinking water ("Direct" group), or DCA for 10-, 32-, or 57-weeks followed by deionized water until sample collection ("Stop" groups). Genome-scaled alterations in DNA methylation were measured by Reduced Representation Bisulfite Sequencing (RRBS) in 78-week liver samples for control, Direct, 10-week Stop DCA exposed mice.
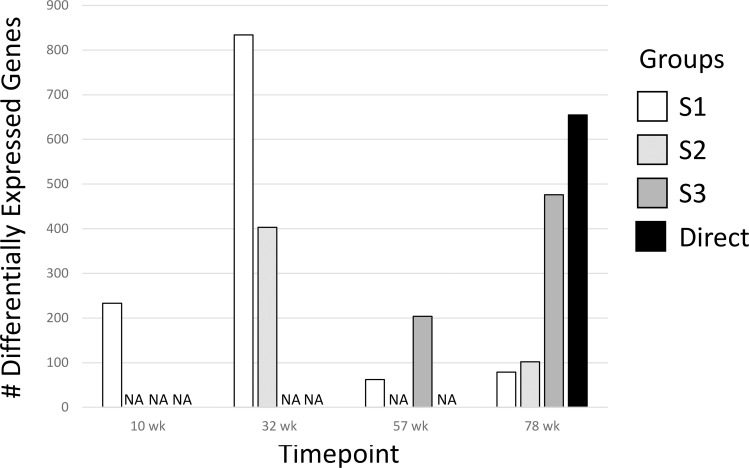
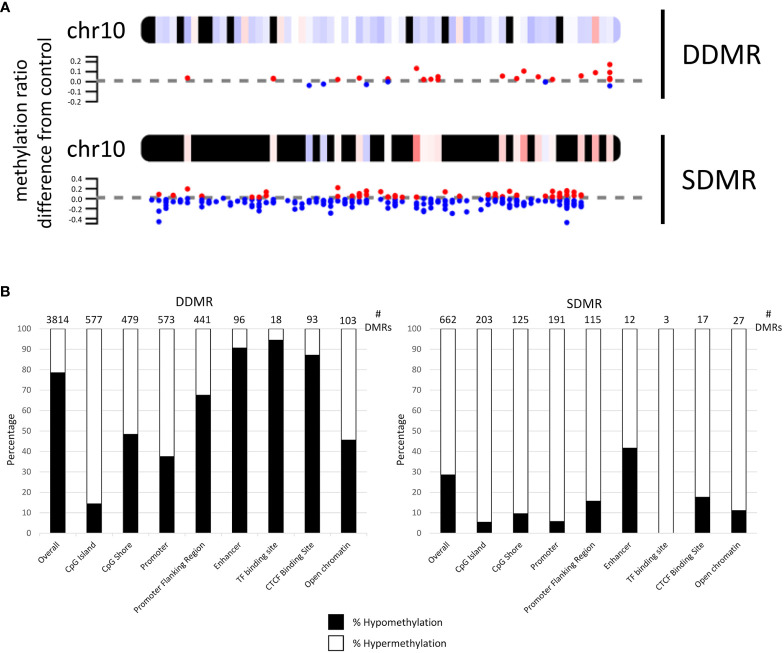
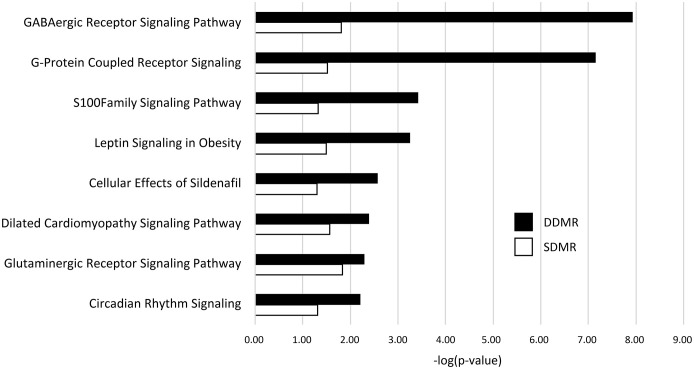
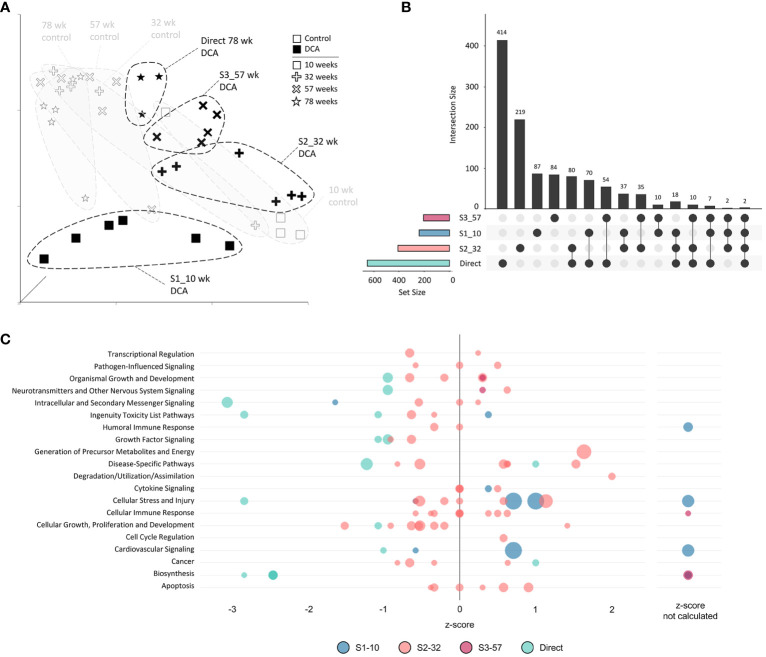
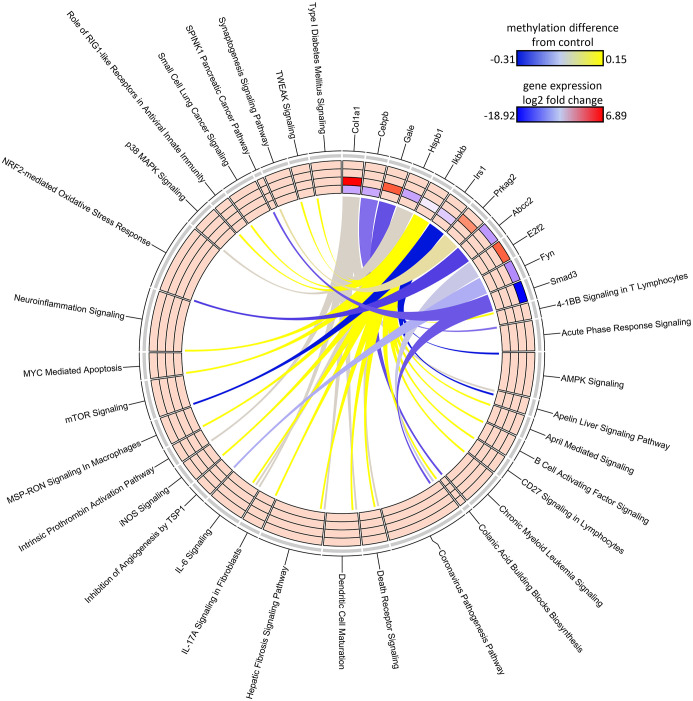
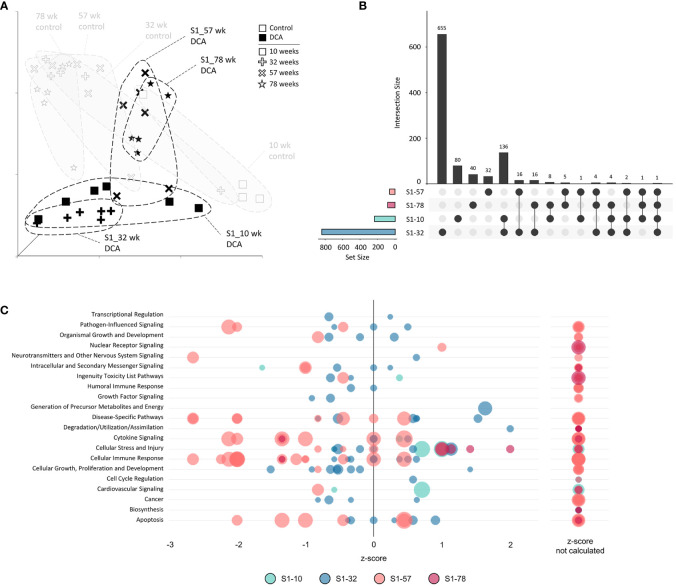
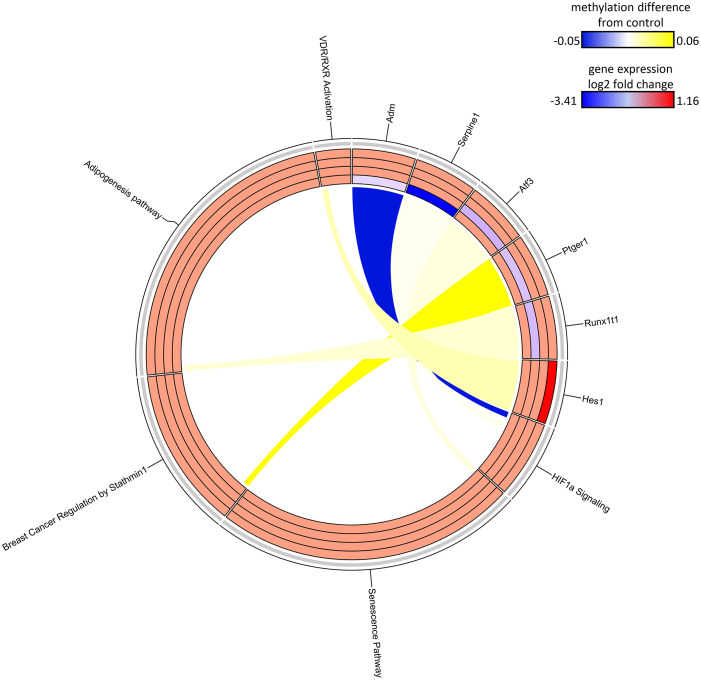
Results: Transcriptomic changes were most robust with concurrent or adjacent timepoints after exposure was withdrawn. We observed a similar pattern with DNA methylation alterations where we noted attenuated differentially methylated regions (DMRs) in the 10-week Stop DCA exposure groups compared to the Direct group at 78-weeks. Gene pathway analysis indicated cellular effects linked to increased oxidative metabolism, a primary mechanism of action for DCA, closer to exposure windows especially early in life. Conversely, many gene signatures and pathways reversed patterns later in life and reflected more pro-tumorigenic patterns for both current and prior DCA exposures. DNA methylation patterns correlated to early gene pathway perturbations, such as cellular signaling, regulation and metabolism, suggesting persistence in the epigenome and possible regulatory effects.
Conclusion: Liver metabolic reprogramming effects of DCA interacted with normal age mechanisms, increasing tumor burden with both continuous and prior DCA exposure in the male B6C3F1 rodent model.
Keywords: DNA methylation (5mC); age; dichloro acetic acid; liver; mouse model; transcriptomics (RNA sequencing); tumorigenesis.
Copyright © 2024 Carswell, Chamberlin, Bennett, Bushel and Chorley.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- EPA . Guidelines for Carcinogen Risk Assessment. Risk Assessment Forum, Washington, D.C: (2005). Available at: https://www.epa.gov/sites/default/files/2013-09/documents/cancer_guideli... (25 April 2024, date last accessed).
LinkOut - more resources
Full Text Sources
Molecular Biology Databases