

This is a preprint.
A Curated Rotamer Library for Common Post-Translational Modifications of Proteins
- PMID: 38764597
- PMCID: PMC11100909
A Curated Rotamer Library for Common Post-Translational Modifications of Proteins
Update in
-
A curated rotamer library for common post-translational modifications of proteins.Bioinformatics. 2024 Jul 1;40(7):btae444. doi: 10.1093/bioinformatics/btae444. Bioinformatics. 2024. PMID: 38995731 Free PMC article.
Abstract
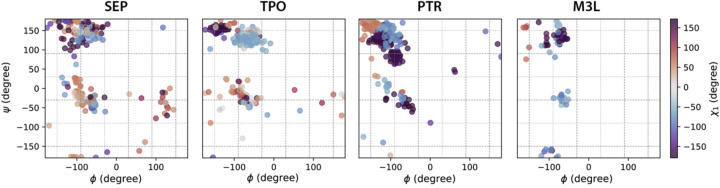
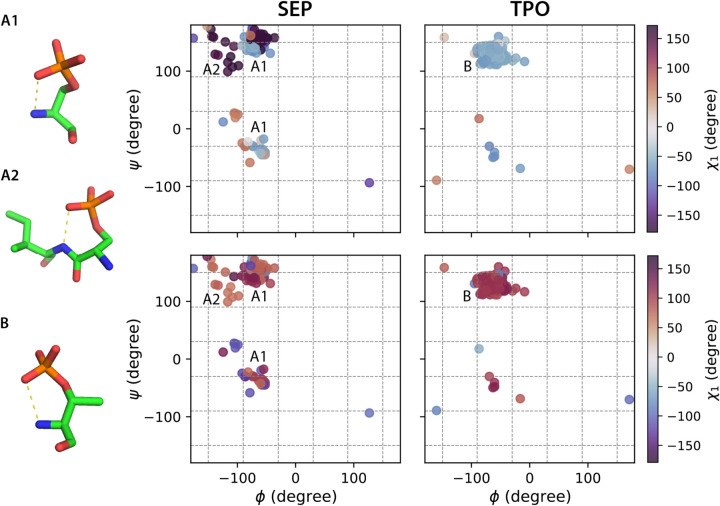
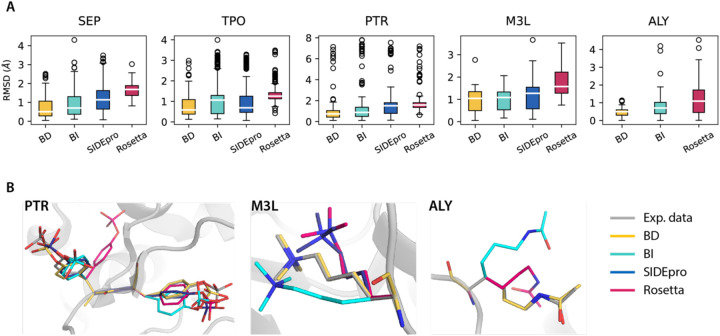
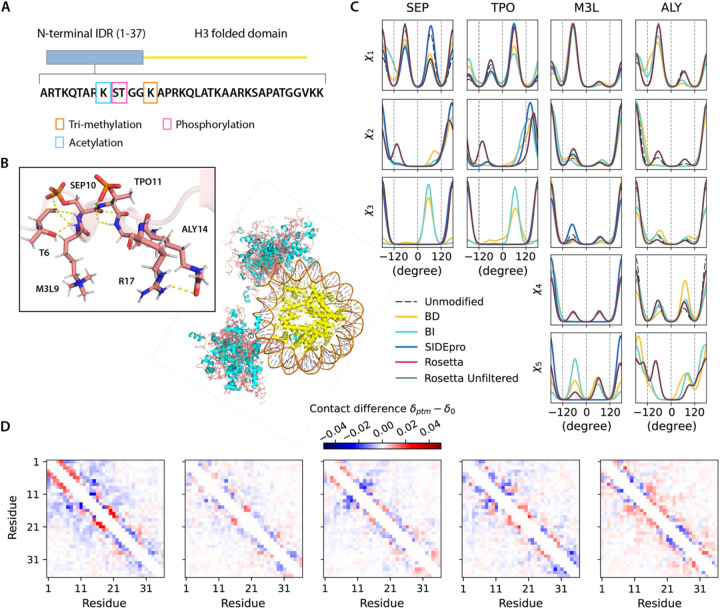
Sidechain rotamer libraries of the common amino acids of a protein are useful for folded protein structure determination and for generating ensembles of intrinsically disordered proteins (IDPs). However much of protein function is modulated beyond the translated sequence through thFiguree introduction of post-translational modifications (PTMs). In this work we have provided a curated set of side chain rotamers for the most common PTMs derived from the RCSB PDB database, including phosphorylated, methylated, and acetylated sidechains. Our rotamer libraries improve upon existing methods such as SIDEpro and Rosetta in predicting the experimental structures for PTMs in folded proteins. In addition, we showcase our PTM libraries in full use by generating ensembles with the Monte Carlo Side Chain Entropy (MCSCE) for folded proteins, and combining MCSCE with the Local Disordered Region Sampling algorithms within IDPConformerGenerator for proteins with intrinsically disordered regions.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous