

Racial inequities and rare CFTR variants: Impact on cystic fibrosis diagnosis and treatment
- PMID: 38765466
- PMCID: PMC11099334
- DOI: 10.1016/j.jcte.2024.100344
Racial inequities and rare CFTR variants: Impact on cystic fibrosis diagnosis and treatment
Abstract
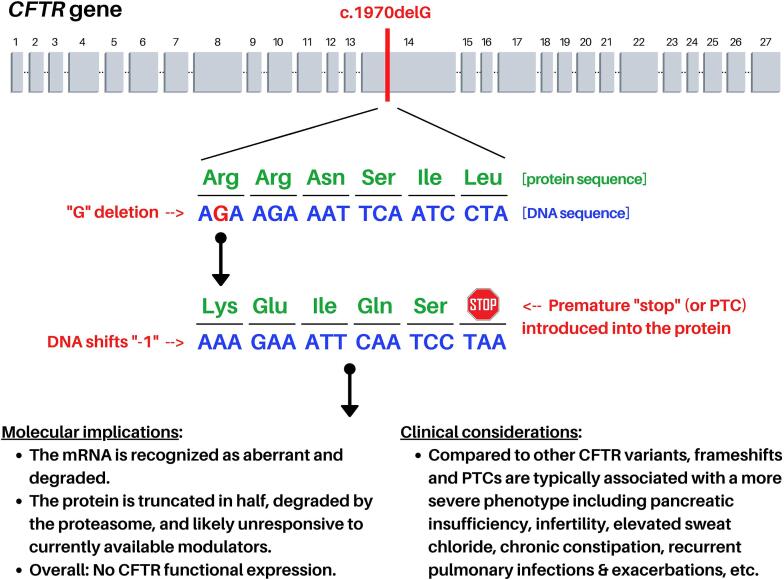
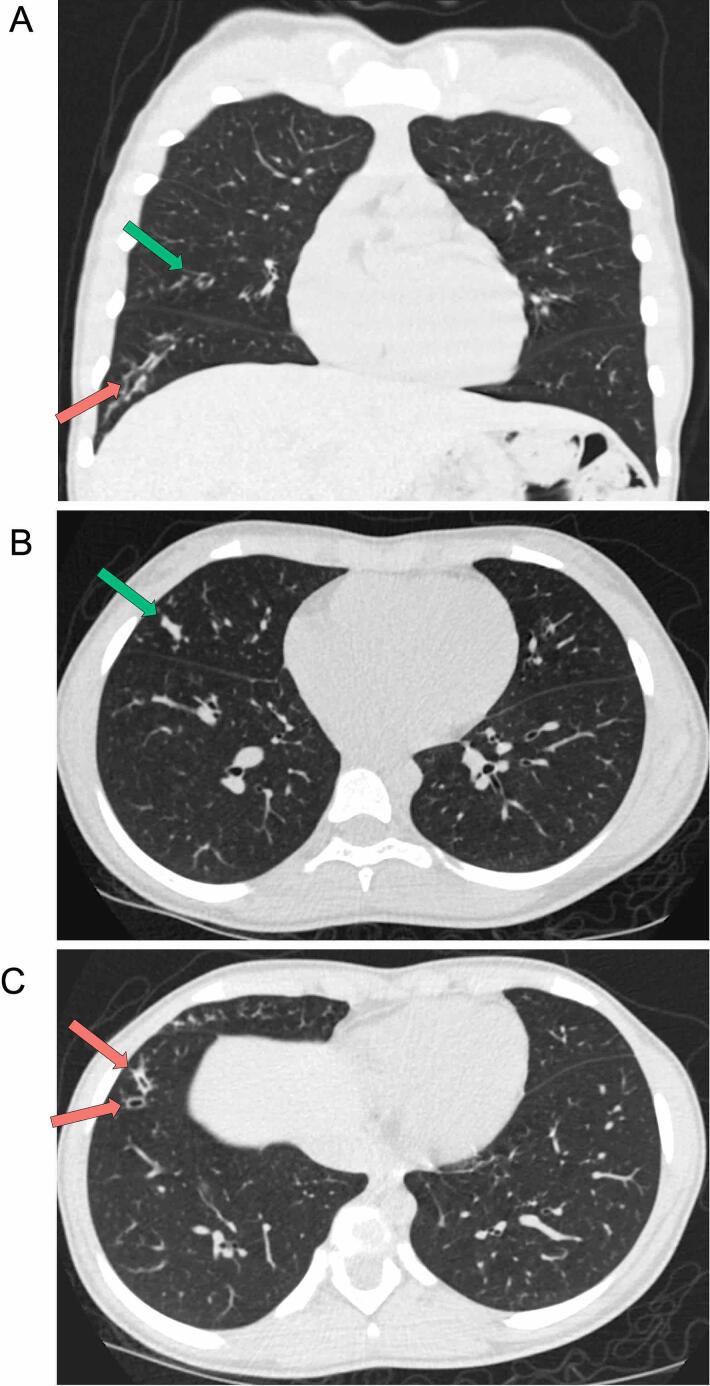
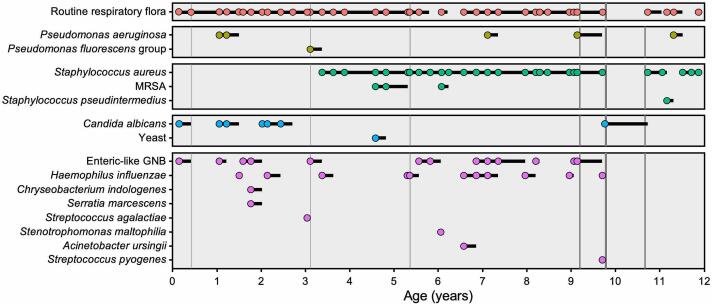
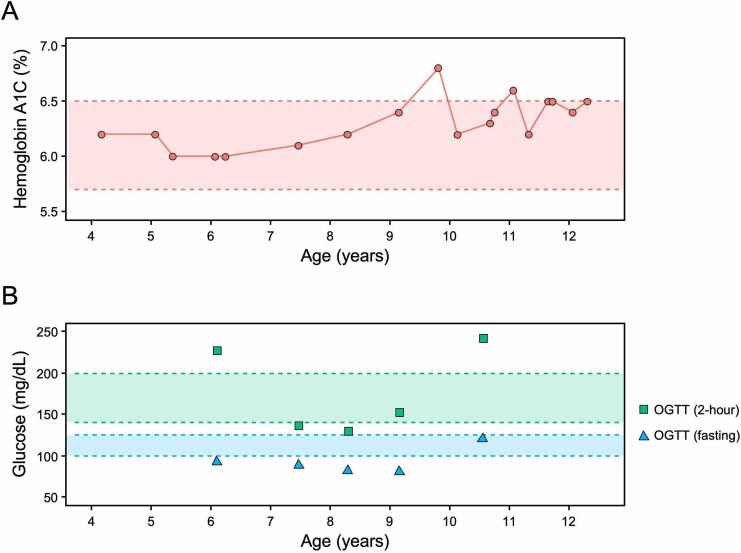
Cystic fibrosis (CF) has been traditionally viewed as a disease that affects White individuals. However, CF occurs among all races, ethnicities, and geographic ancestries. The disorder results from mutations in the CF transmembrane conductance regulator (CFTR). Varying incidence of CF is reported among Black, Indigenous, and People of Color (BIPOC), who typically exhibit worse clinical outcomes. These populations are more likely to carry rare CFTR variants omitted from newborn screening panels, leading to disparities in care such as delayed diagnosis and treatment. In this study, we present a case-in-point describing an individual of Gambian descent identified with CF. Patient genotype includes a premature termination codon (PTC) (c.2353C>T) and previously undescribed single nucleotide deletion (c.1970delG), arguing against effectiveness of currently available CFTR modulator-based interventions. Strategies for overcoming these two variants will likely include combinations of PTC suppressors, nonsense mediated decay inhibitors, and/or alternative approaches (e.g. gene therapy). Investigations such as the present study establish a foundation from which therapeutic treatments may be developed. Importantly, c.2353C>T and c.1970delG were not detected in the patient by traditional CFTR screening panels, which include an implicit racial and ethnic diagnostic bias as these tests are comprised of mutations largely observed in people of European ancestry. We suggest that next-generation sequencing of CFTR should be utilized to confirm or exclude a CF diagnosis, in order to equitably serve BIPOC individuals. Additional epidemiologic data, basic science investigations, and translational work are imperative for improving understanding of disease prevalence and progression, CFTR variant frequency, genotype-phenotype correlation, pharmacologic responsiveness, and personalized medicine approaches for patients with African ancestry and other historically understudied geographic lineages.
Keywords: African; Cystic fibrosis; Cystic fibrosis-related diabetes; Diagnosis; Genetics; Newborn screening.
© 2024 The Author(s).
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
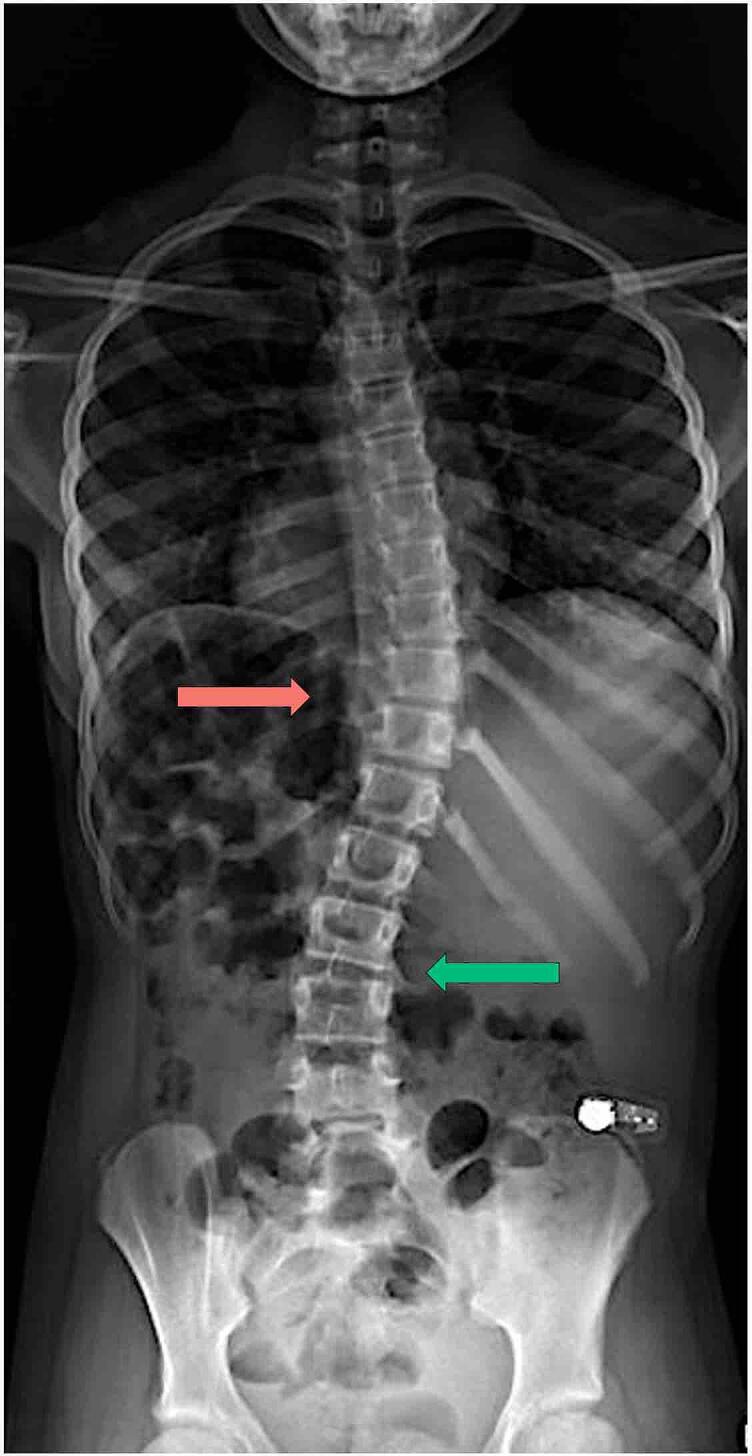
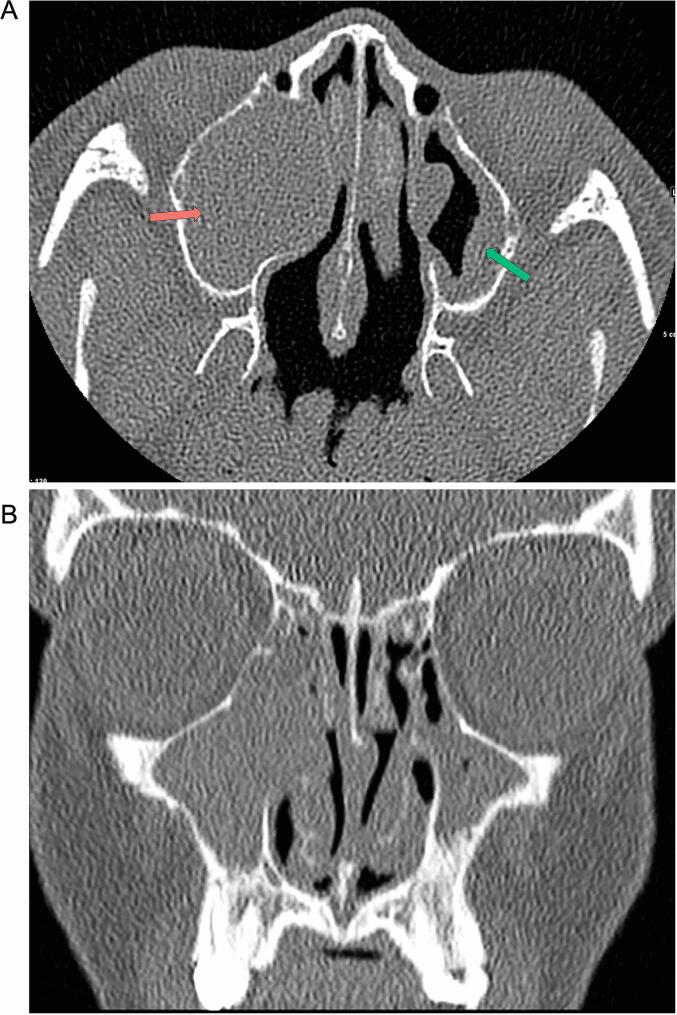
References
-
- Stalvey M.S., Clines G.A. Cystic fibrosis-related bone disease: insights into a growing problem. Curr Opin Endocrinol Diabetes Obes. 2013;20:547–552. doi: 10.1097/01.med.0000436191.87727.ec. - DOI - PMC - PubMed
-
- Walkowiak J., Herzig K.H., Witt M., Pogorzelski A., Piotrowski R., Barra E., et al. Analysis of exocrine pancreatic function in cystic fibrosis: one mild CFTR mutation does not exclude pancreatic insufficiency. Eur J Clin Invest. 2001;31:796–801. doi: 10.1046/j.1365-2362.2001.00876.x. - DOI - PubMed
-
- Moran A., Brunzell C., Cohen R.C., Katz M., Marshall B.C., Onady G., et al. Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care. 2010;33:2697–2708. doi: 10.2337/dc10-1768. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
