

This is a preprint.
De novo gene synthesis by an antiviral reverse transcriptase
- PMID: 38766058
- PMCID: PMC11100668
- DOI: 10.1101/2024.05.08.593200
De novo gene synthesis by an antiviral reverse transcriptase
Update in
-
De novo gene synthesis by an antiviral reverse transcriptase.Science. 2024 Oct 4;386(6717):eadq0876. doi: 10.1126/science.adq0876. Epub 2024 Oct 4. Science. 2024. PMID: 39116258 Free PMC article.
Abstract
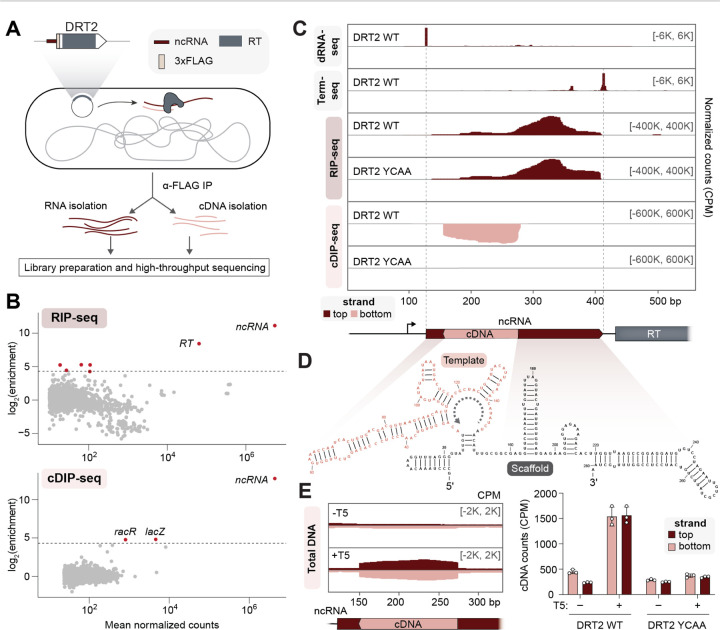
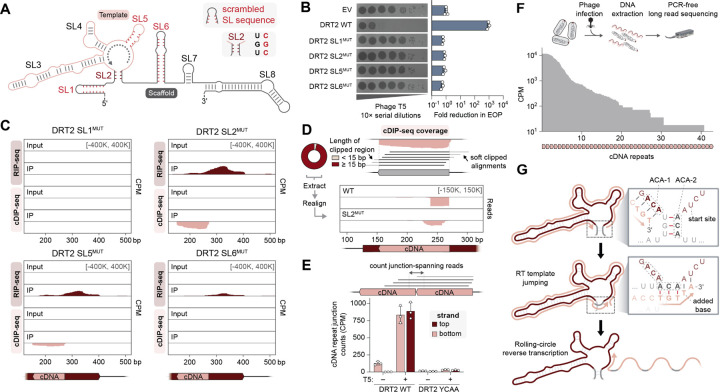
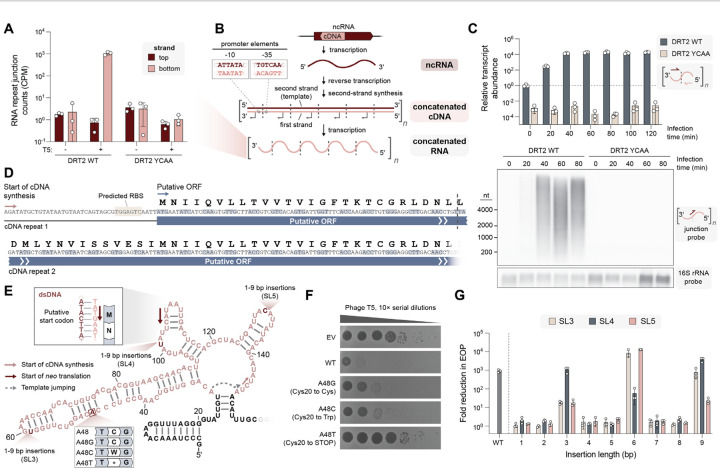
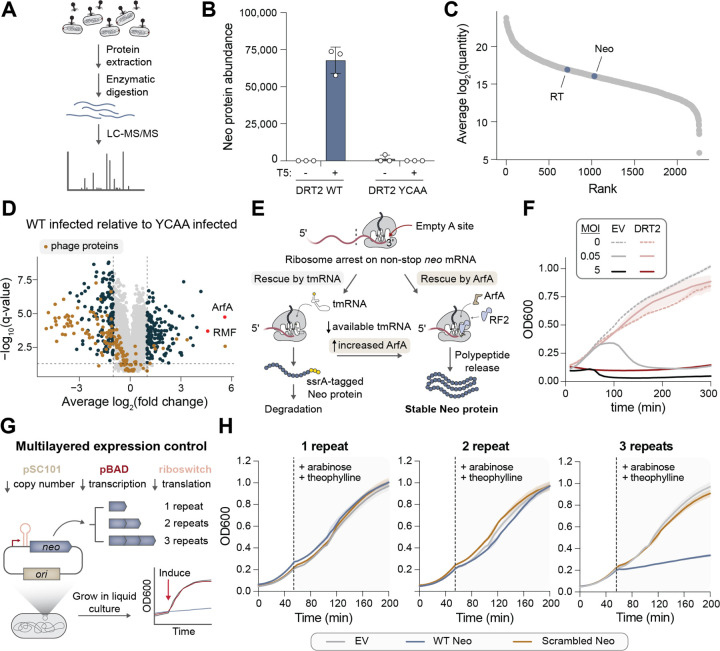
Bacteria defend themselves from viral infection using diverse immune systems, many of which sense and target foreign nucleic acids. Defense-associated reverse transcriptase (DRT) systems provide an intriguing counterpoint to this immune strategy by instead leveraging DNA synthesis, but the identities and functions of their DNA products remain largely unknown. Here we show that DRT2 systems execute an unprecedented immunity mechanism that involves de novo gene synthesis via rolling-circle reverse transcription of a non-coding RNA (ncRNA). Unbiased profiling of RT-associated RNA and DNA ligands in DRT2-expressing cells revealed that reverse transcription generates concatenated cDNA repeats through programmed template jumping on the ncRNA. The presence of phage then triggers second-strand cDNA synthesis, leading to the production of long double-stranded DNA. Remarkably, this DNA product is efficiently transcribed, generating messenger RNAs that encode a stop codon-less, never-ending ORF (neo) whose translation causes potent growth arrest. Phylogenetic analyses and screening of diverse DRT2 homologs further revealed broad conservation of rolling-circle reverse transcription and Neo protein function. Our work highlights an elegant expansion of genome coding potential through RNA-templated gene creation, and challenges conventional paradigms of genetic information encoded along the one-dimensional axis of genomic DNA.
Conflict of interest statement
Columbia University has filed a patent application related to this work. S.H.S. is a co-founder and scientific advisor to Dahlia Biosciences, a scientific advisor to CrisprBits and Prime Medicine, and an equity holder in Dahlia Biosciences and CrisprBits.
Figures
References
-
- Frost L. S., Leplae R., Summers A. O., Toussaint A., Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol 3, 722–732 (2005). - PubMed
-
- Canapa A., Barucca M., Biscotti M. A., Forconi M., Olmo E., Transposons, Genome Size, and Evolutionary Insights in Animals. Cytogenetic and Genome Research 147, 217–239 (2016). - PubMed
-
- Koonin E. V., Makarova K. S., Wolf Y. I., Krupovic M., Evolutionary entanglement of mobile genetic elements and host defence systems: guns for hire. Nat Rev Genet 21, 119–131 (2020). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous