

This is a preprint.
Virtual Screening of Molecules via Neural Fingerprint-based Deep Learning Technique
- PMID: 38766198
- PMCID: PMC11100899
- DOI: 10.21203/rs.3.rs-4355625/v1
Virtual Screening of Molecules via Neural Fingerprint-based Deep Learning Technique
Update in
-
Efficient and Explainable Virtual Screening of Molecules through Fingerprint-Generating Networks Integrated with Artificial Neural Networks.ACS Omega. 2025 Jan 28;10(5):4896-4911. doi: 10.1021/acsomega.4c10289. eCollection 2025 Feb 11. ACS Omega. 2025. PMID: 39959102 Free PMC article.
Abstract
A machine learning-based drug screening technique has been developed and optimized using convolutional neural network-derived fingerprints. The optimization of weights in the neural network-based fingerprinting technique was compared with fixed Morgan fingerprints in regard to binary classification on drug-target binding affinity. The assessment was carried out using six different target proteins using randomly chosen small molecules from the ZINC15 database for training. This new architecture proved to be more efficient in screening molecules that less favorably bind to specific targets and retaining molecules that favorably bind to it.
Scientific contribution: We have developed a new neural fingerprint-based screening model that has a significant ability to capture hits. Despite using a smaller dataset, this model is capable of mapping chemical space similar to other contemporary algorithms designed for molecular screening. The novelty of the present algorithm lies in the speed with which the models are trained and tuned before testing its predictive capabilities and hence is a significant step forward in the field of machine learning-embedded computational drug discovery.
Keywords: Artificial neural network; deep learning; drug design; machine learning; neural network-based fingerprinting; structure-based inhibitor design.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures
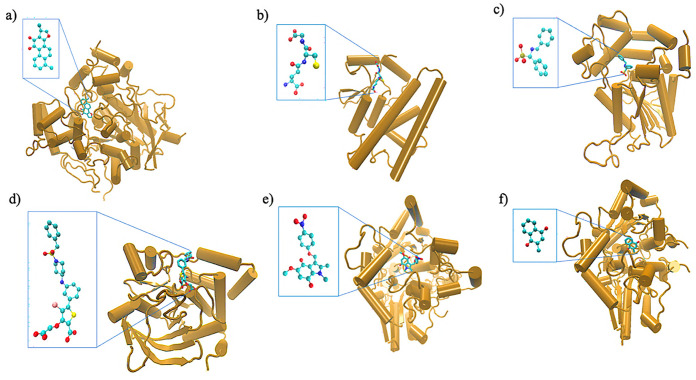
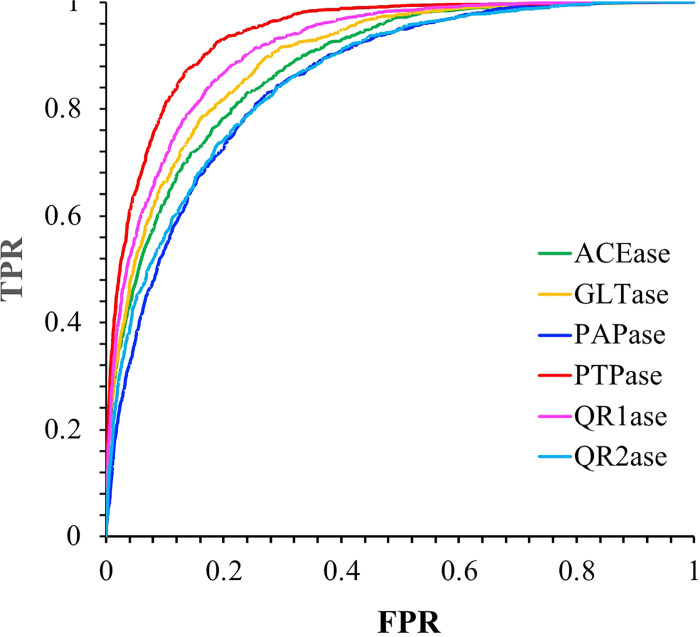
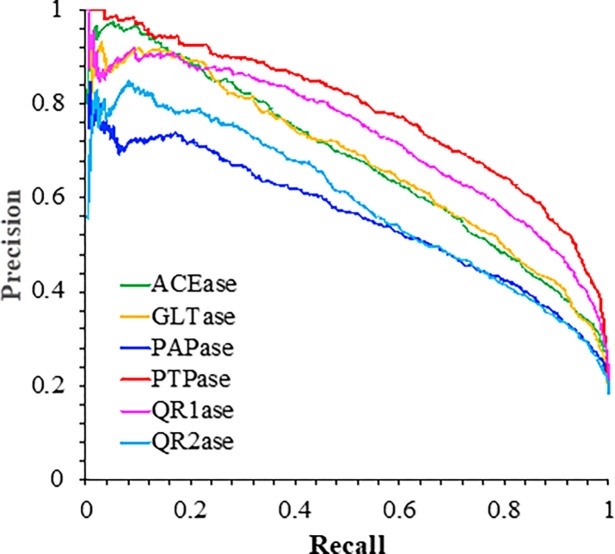
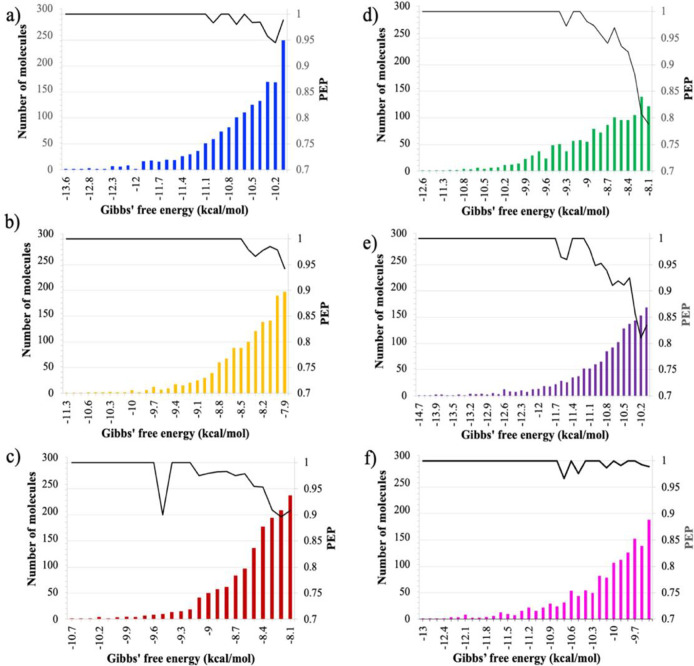

References
-
- Bajorath J (2022) Deep Machine Learning for Computer-Aided Drug Design. Front Drug Discov 2:829043. 10.3389/fddsv.2022.829043 - DOI
-
- Kuntz D, Wilson AK (2022) Machine learning, artificial intelligence, and chemistry: How smart algorithms are reshaping simulation and the laboratory. Pure Appl Chem 94:1019–1054. 10.1515/pac-2022-0202 - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
