

In vitro reconstitution of epigenetic reprogramming in the human germ line
- PMID: 38768632
- PMCID: PMC11222161
- DOI: 10.1038/s41586-024-07526-6
In vitro reconstitution of epigenetic reprogramming in the human germ line
Abstract
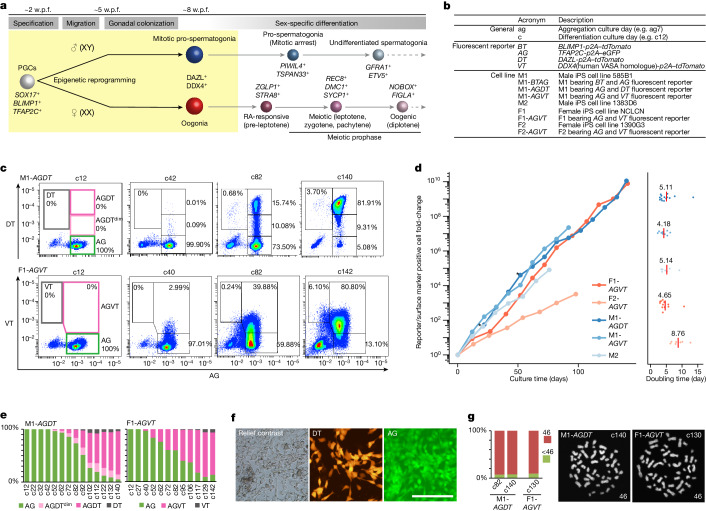
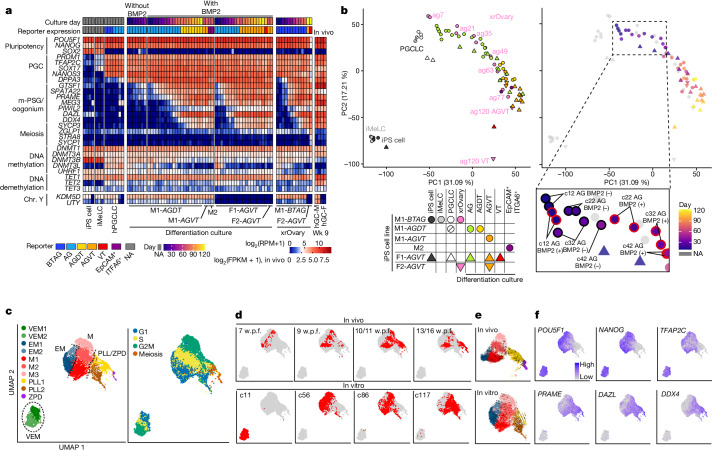
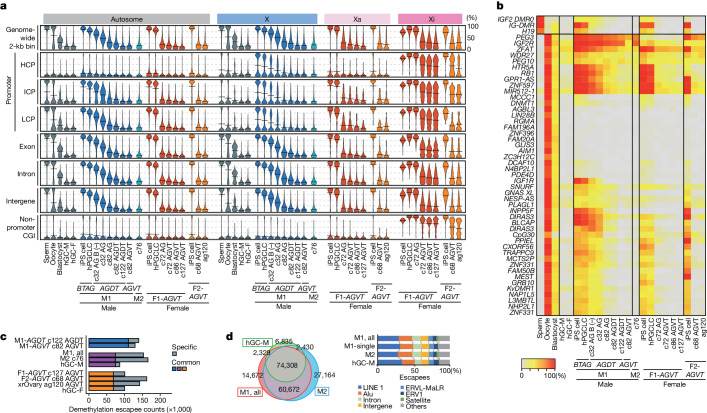
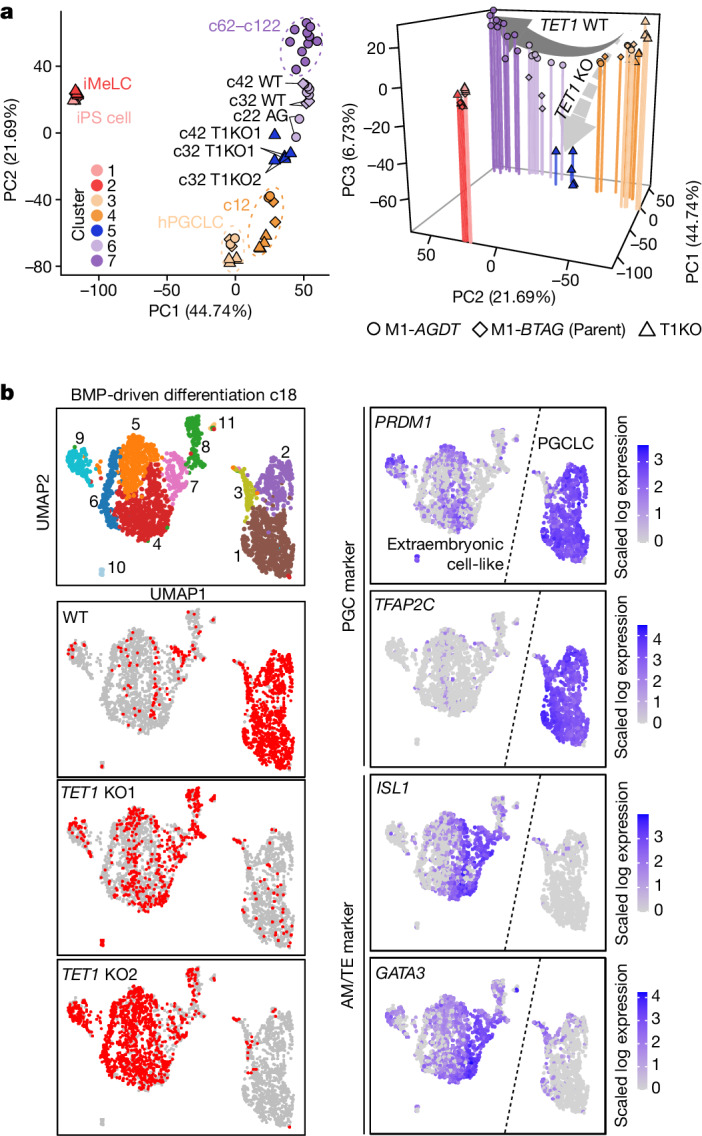
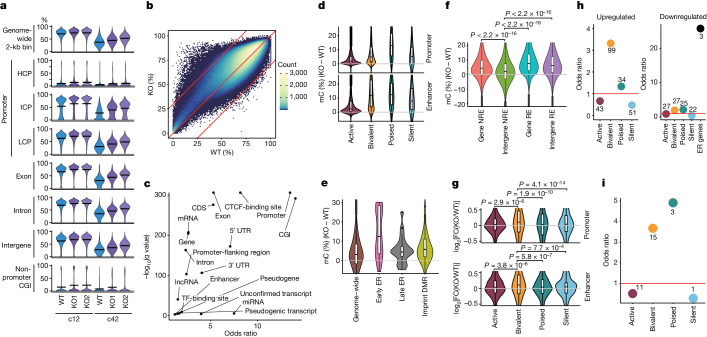
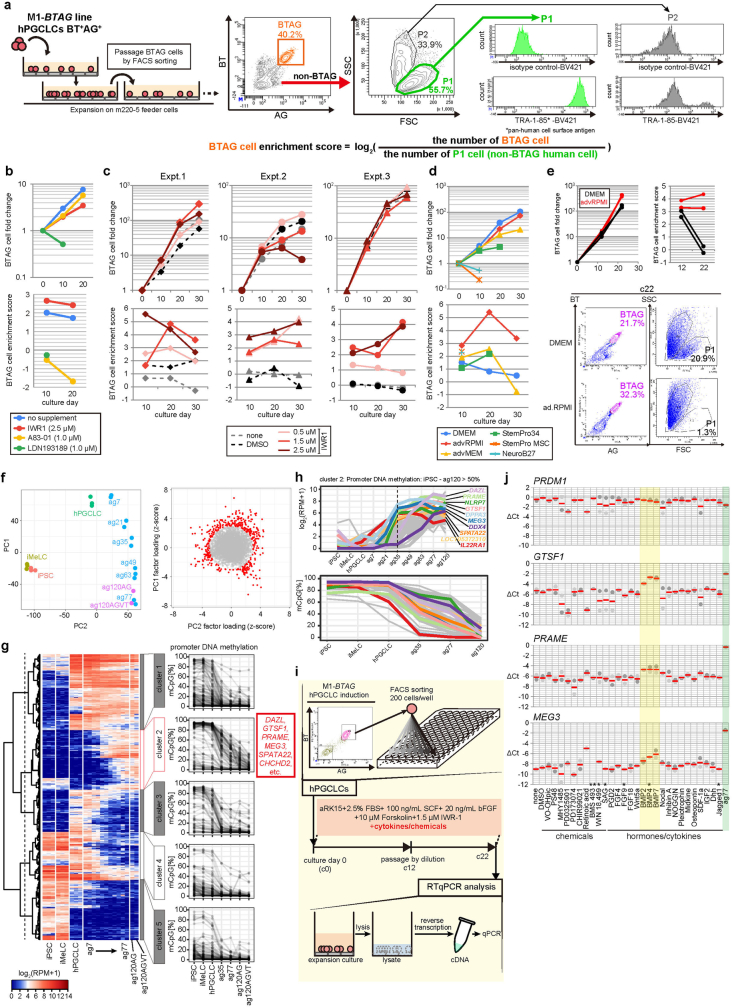
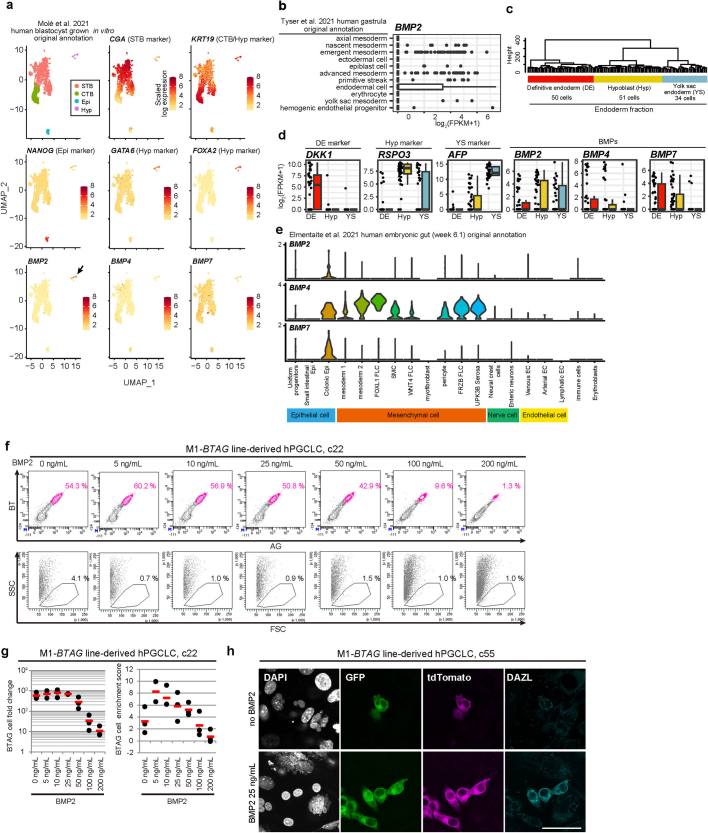
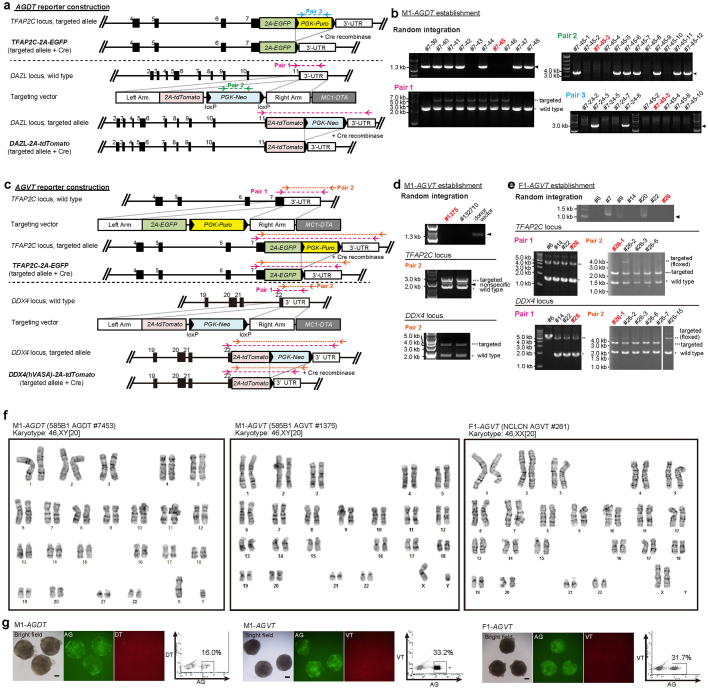
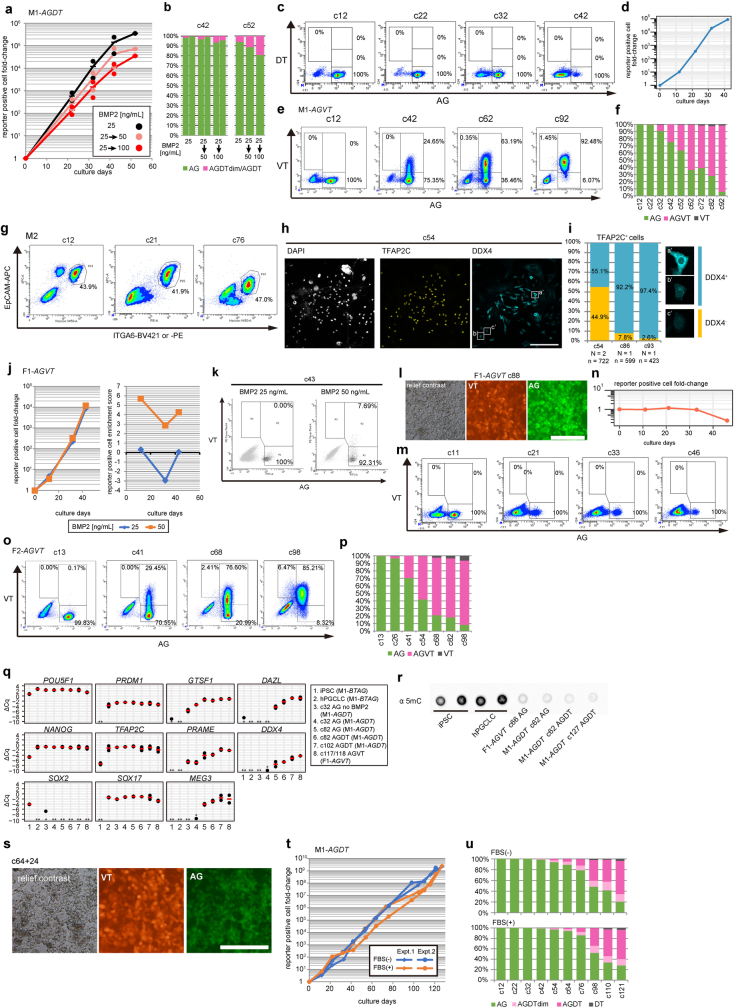
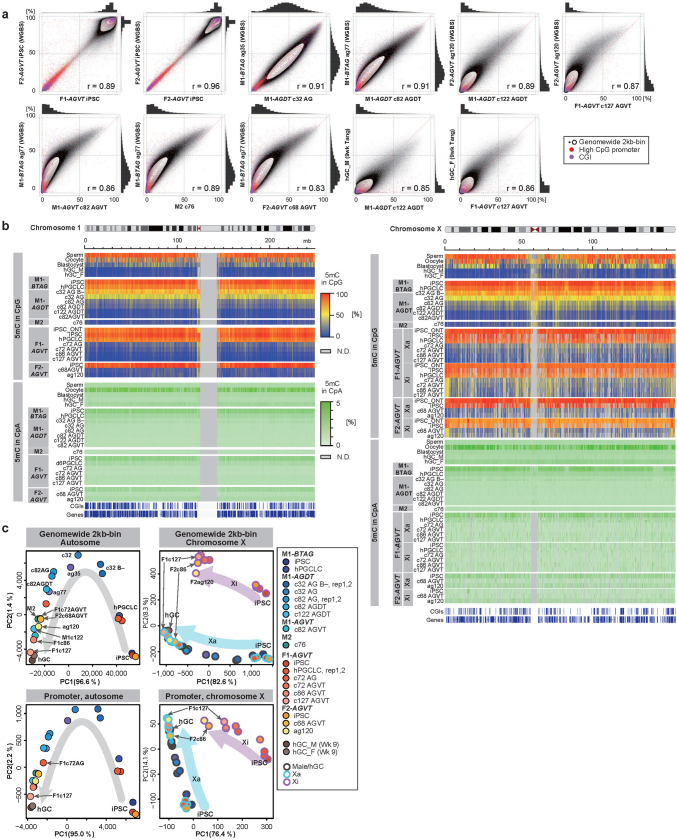
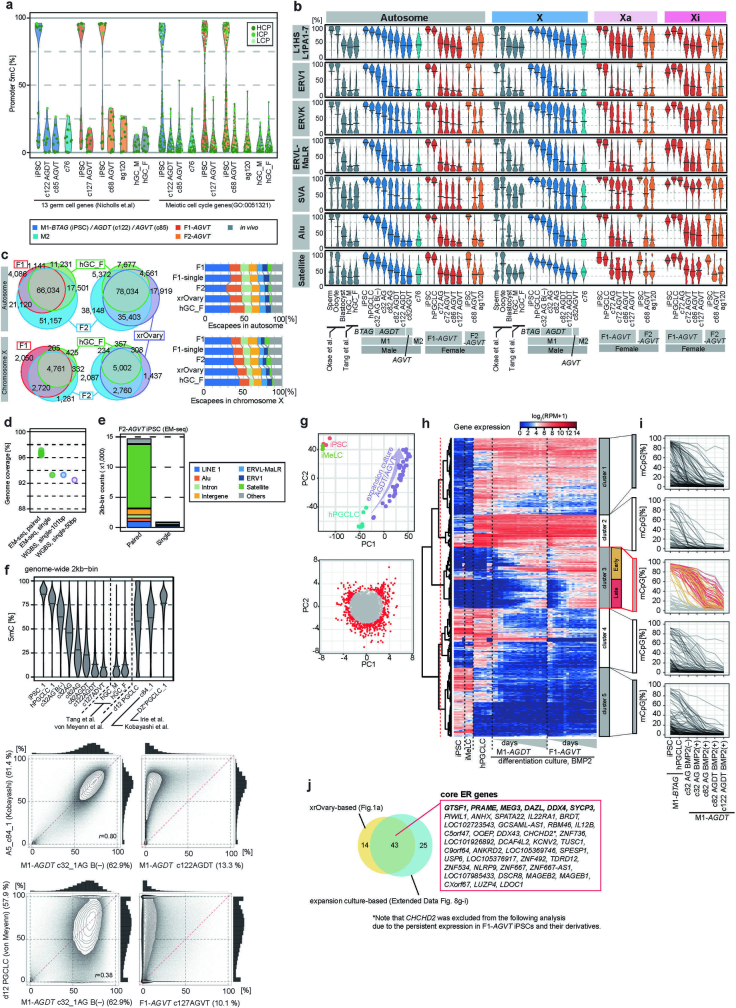
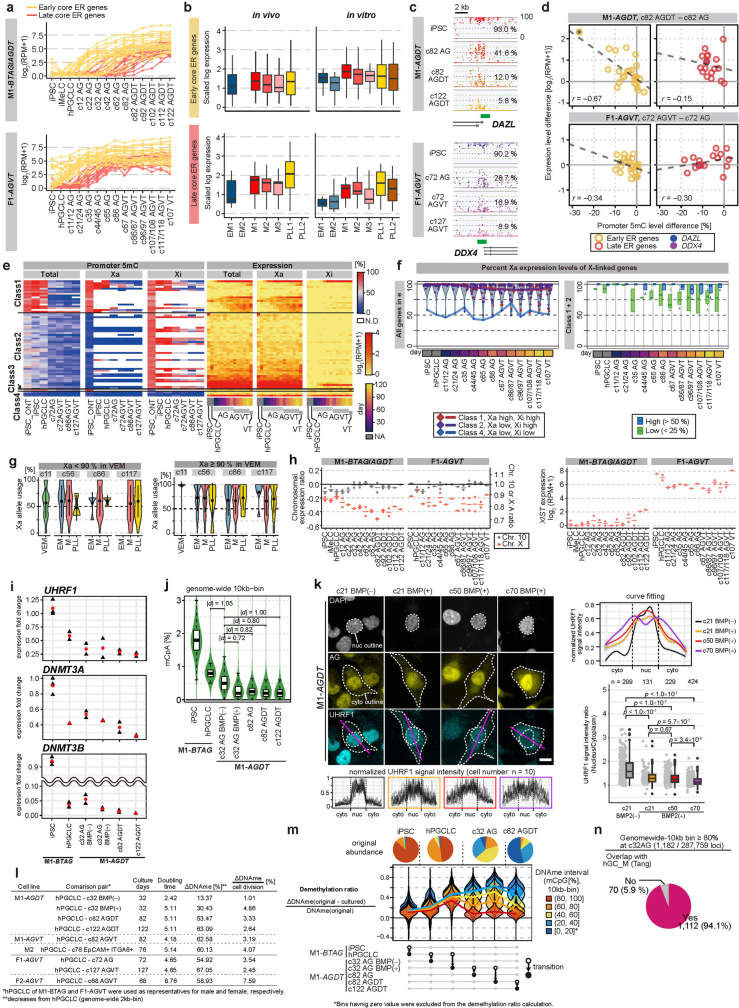
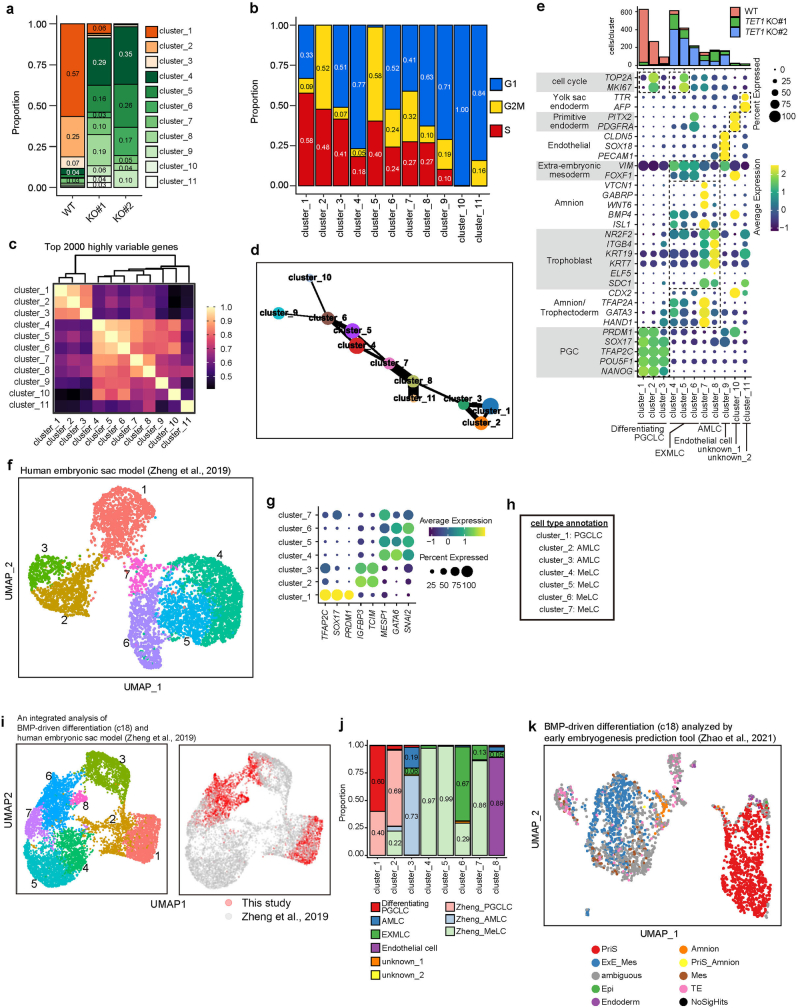
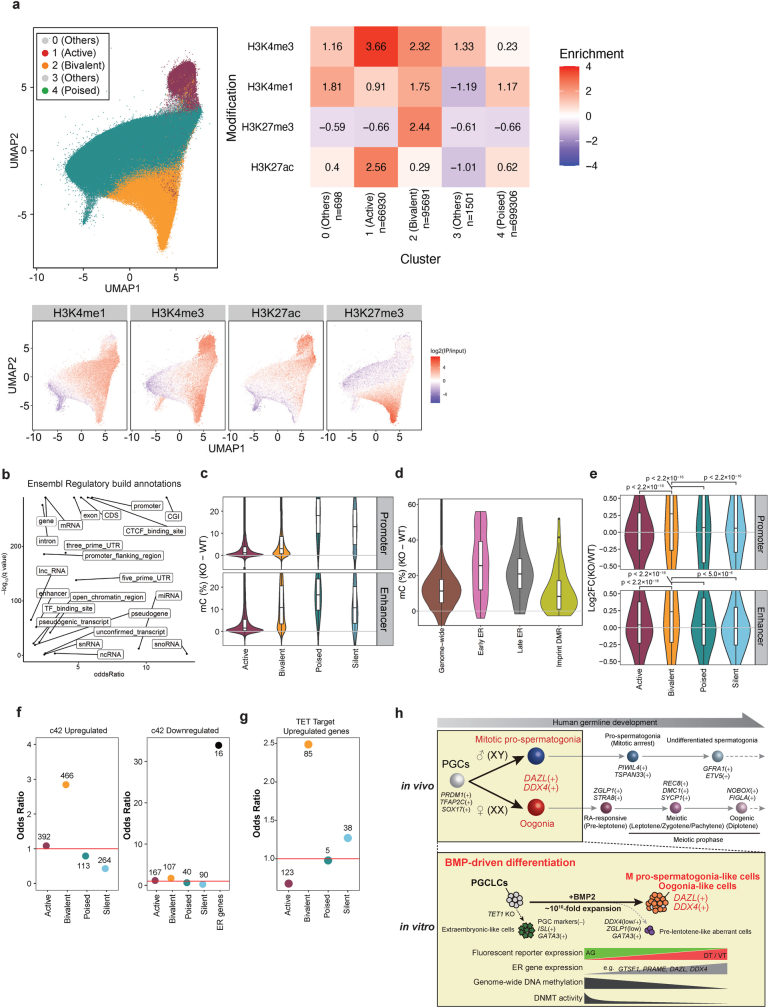
Epigenetic reprogramming resets parental epigenetic memories and differentiates primordial germ cells (PGCs) into mitotic pro-spermatogonia or oogonia. This process ensures sexually dimorphic germ cell development for totipotency1. In vitro reconstitution of epigenetic reprogramming in humans remains a fundamental challenge. Here we establish a strategy for inducing epigenetic reprogramming and differentiation of pluripotent stem-cell-derived human PGC-like cells (hPGCLCs) into mitotic pro-spermatogonia or oogonia, coupled with their extensive amplification (about >1010-fold). Bone morphogenetic protein (BMP) signalling is a key driver of these processes. BMP-driven hPGCLC differentiation involves attenuation of the MAPK (ERK) pathway and both de novo and maintenance DNA methyltransferase activities, which probably promote replication-coupled, passive DNA demethylation. hPGCLCs deficient in TET1, an active DNA demethylase abundant in human germ cells2,3, differentiate into extraembryonic cells, including amnion, with de-repression of key genes that bear bivalent promoters. These cells fail to fully activate genes vital for spermatogenesis and oogenesis, and their promoters remain methylated. Our study provides a framework for epigenetic reprogramming in humans and an important advance in human biology. Through the generation of abundant mitotic pro-spermatogonia and oogonia-like cells, our results also represent a milestone for human in vitro gametogenesis research and its potential translation into reproductive medicine.
© 2024. The Author(s).
Conflict of interest statement
M.S., Y.M. and R.Y., together with Kyoto University, have filed a provisional patent application (2023-133928) covering the propagation and differentiation of germ cells induced from human PS cells. All other authors declare no competing interests.
Figures



Comment in
-
A new signalling-based system for germ cell reprogramming.Nat Rev Urol. 2024 Jul;21(7):390. doi: 10.1038/s41585-024-00907-6. Nat Rev Urol. 2024. PMID: 38872010 No abstract available.
References
-
- Tang WW, Kobayashi T, Irie N, Dietmann S, Surani MA. Specification and epigenetic programming of the human germ line. Nat. Rev. Genet. 2016;17:585–600. - PubMed
-
- Hertig AT, et al. A thirteen-day human ovum studied histochemically. Am. J. Obstet. Gynecol. 1958;76:1025–1040. - PubMed
-
- Sasaki K, et al. The germ cell fate of cynomolgus monkeys is specified in the nascent amnion. Dev. Cell. 2016;39:169–185. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
