

A deep catalogue of protein-coding variation in 983,578 individuals
- PMID: 38768635
- PMCID: PMC11254753
- DOI: 10.1038/s41586-024-07556-0
A deep catalogue of protein-coding variation in 983,578 individuals
Erratum in
-
Author Correction: A deep catalogue of protein-coding variation in 983,578 individuals.Nature. 2025 Jan;637(8047):E26. doi: 10.1038/s41586-024-08571-x. Nature. 2025. PMID: 39779867 Free PMC article. No abstract available.
Abstract
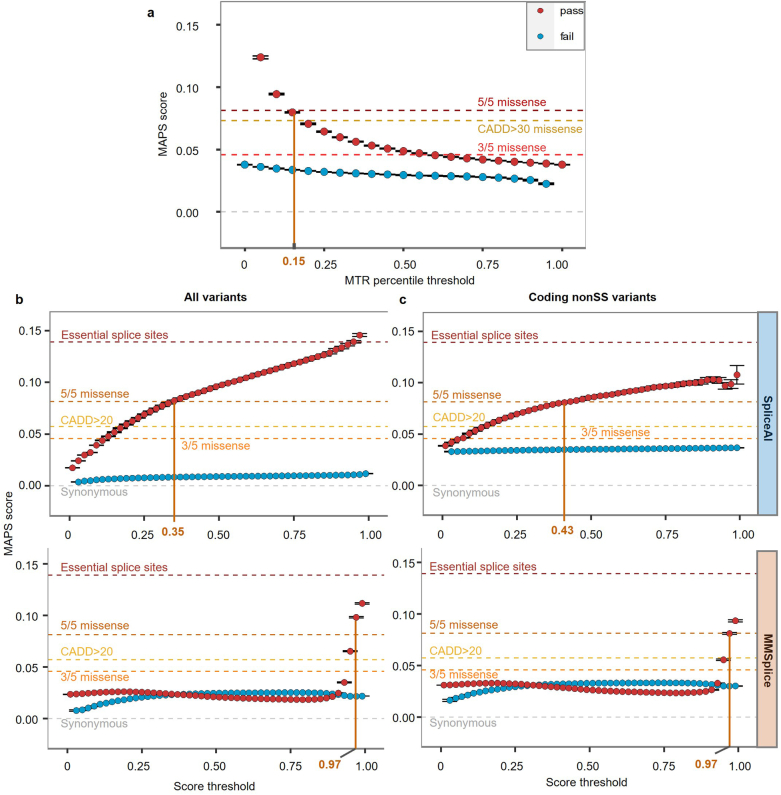
Rare coding variants that substantially affect function provide insights into the biology of a gene1-3. However, ascertaining the frequency of such variants requires large sample sizes4-8. Here we present a catalogue of human protein-coding variation, derived from exome sequencing of 983,578 individuals across diverse populations. In total, 23% of the Regeneron Genetics Center Million Exome (RGC-ME) data come from individuals of African, East Asian, Indigenous American, Middle Eastern and South Asian ancestry. The catalogue includes more than 10.4 million missense and 1.1 million predicted loss-of-function (pLOF) variants. We identify individuals with rare biallelic pLOF variants in 4,848 genes, 1,751 of which have not been previously reported. From precise quantitative estimates of selection against heterozygous loss of function (LOF), we identify 3,988 LOF-intolerant genes, including 86 that were previously assessed as tolerant and 1,153 that lack established disease annotation. We also define regions of missense depletion at high resolution. Notably, 1,482 genes have regions that are depleted of missense variants despite being tolerant of pLOF variants. Finally, we estimate that 3% of individuals have a clinically actionable genetic variant, and that 11,773 variants reported in ClinVar with unknown significance are likely to be deleterious cryptic splice sites. To facilitate variant interpretation and genetics-informed precision medicine, we make this resource of coding variation from the RGC-ME dataset publicly accessible through a variant allele frequency browser.
© 2024. The Author(s).
Conflict of interest statement
K.Y.S., X.B., S.C., S. Bao, C.Z., M.K., J. Backman, T.J., E.M., G.M., A.G., A. Mansfield, B.B., S.G., L.H., A. Marcketta, A.E.L., L.G., A.H., M.D.K., D.S., J.S., J. Bovijn, S.G., A.D.G., V.M.R., A.L., J.R.V., M.C., T.T., J.D.O., A.R.S., M.L.C., M.N., A.B., G.A., J.M., J.G.R., W.S. and S. Balasubramanian are current employees and/or stockholders of Regeneron Genetics Center or Regeneron Pharmaceuticals. H.M.K. was a former employee of Regeneron Genetics Center.
Figures













References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
