

Upregulation of the AMPK-FOXO1-PDK4 pathway is a primary mechanism of pyruvate dehydrogenase activity reduction in tafazzin-deficient cells
- PMID: 38769106
- PMCID: PMC11106297
- DOI: 10.1038/s41598-024-62262-1
Upregulation of the AMPK-FOXO1-PDK4 pathway is a primary mechanism of pyruvate dehydrogenase activity reduction in tafazzin-deficient cells
Erratum in
-
Author Correction: Upregulation of the AMPK-FOXO1-PDK4 pathway is a primary mechanism of pyruvate dehydrogenase activity reduction in tafazzin-deficient cells.Sci Rep. 2024 Jun 25;14(1):14633. doi: 10.1038/s41598-024-65477-4. Sci Rep. 2024. PMID: 38918568 Free PMC article. No abstract available.
Abstract
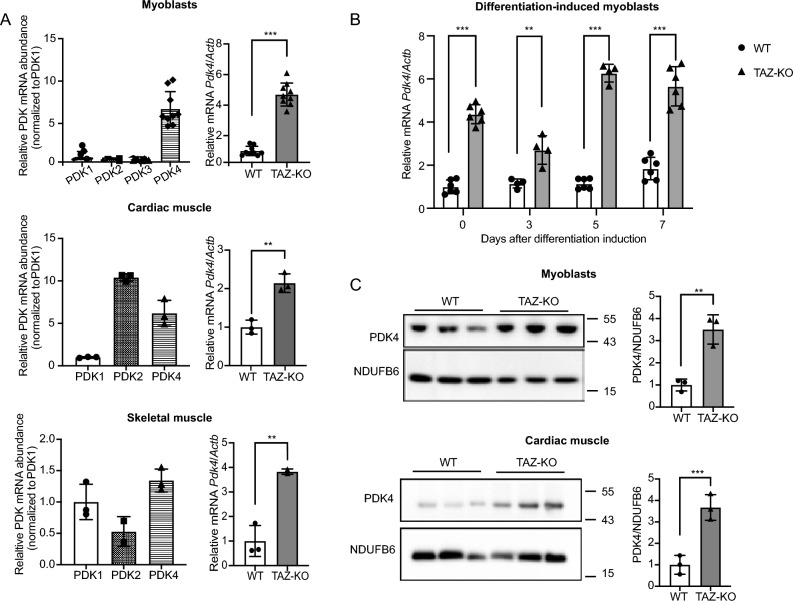
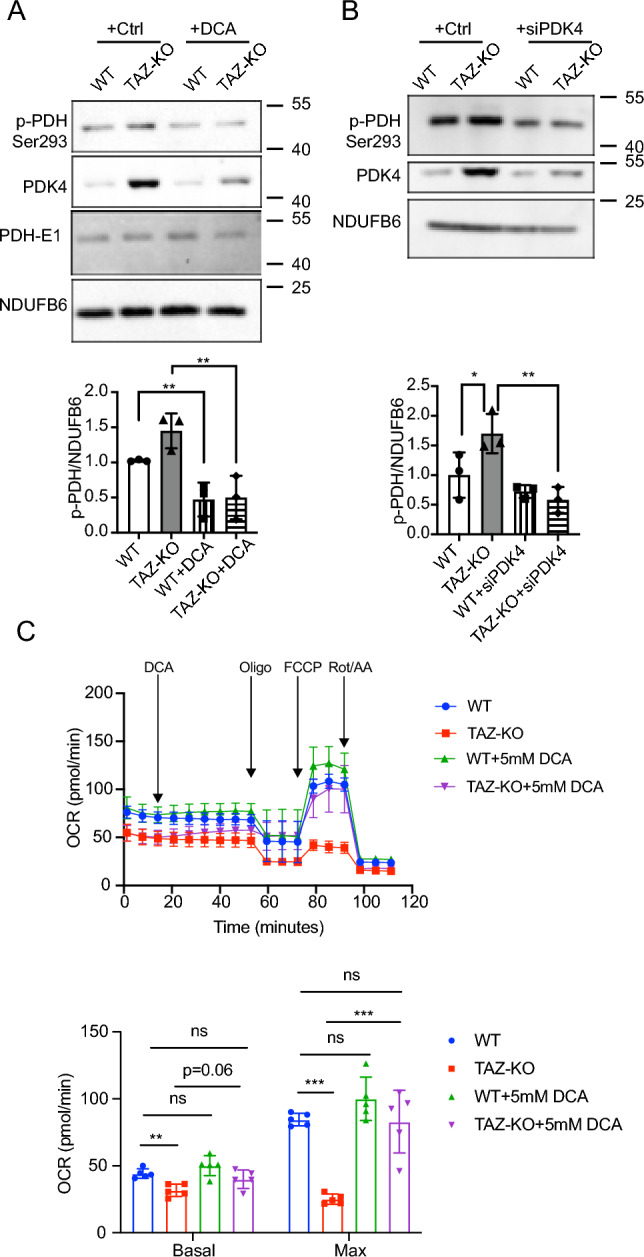
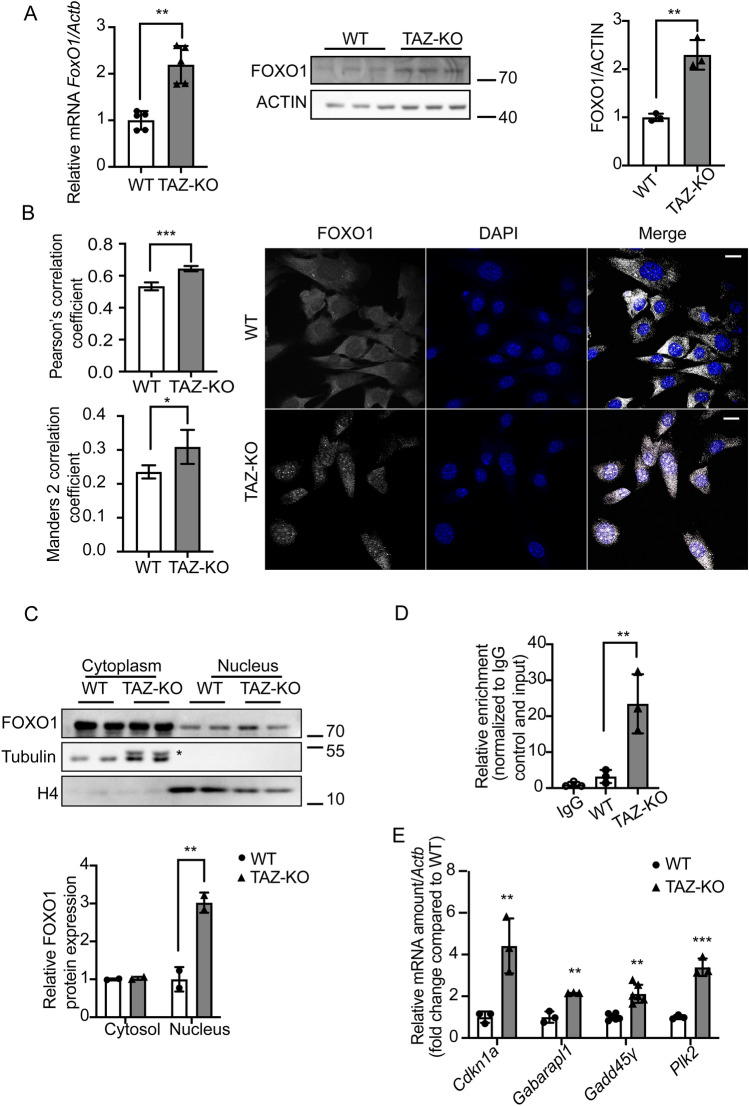
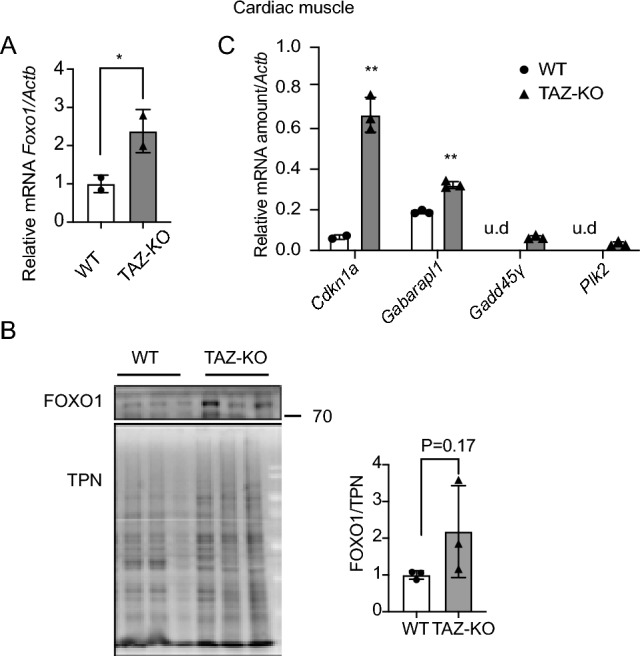
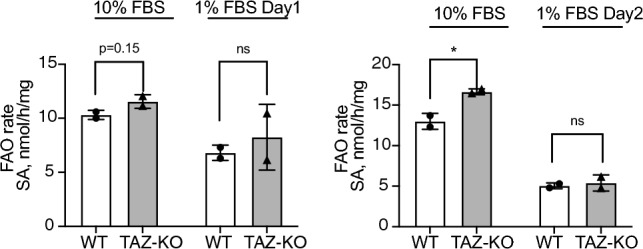
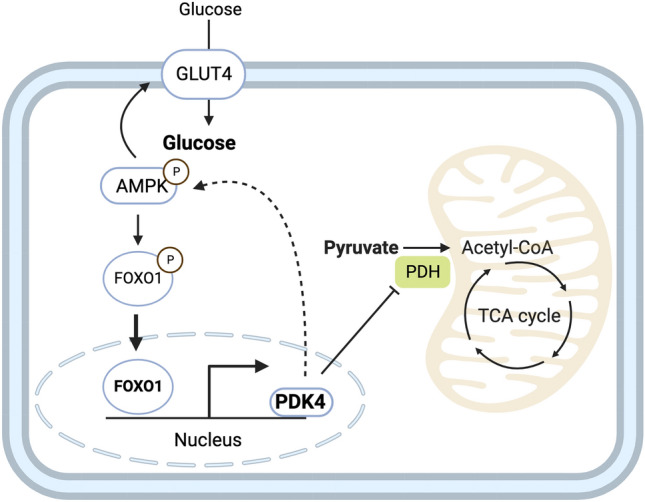
Barth syndrome (BTHS) is a rare disorder caused by mutations in the TAFAZZIN gene. Previous studies from both patients and model systems have established metabolic dysregulation as a core component of BTHS pathology. In particular, features such as lactic acidosis, pyruvate dehydrogenase (PDH) deficiency, and aberrant fatty acid and glucose oxidation have been identified. However, the lack of a mechanistic understanding of what causes these conditions in the context of BTHS remains a significant knowledge gap, and this has hindered the development of effective therapeutic strategies for treating the associated metabolic problems. In the current study, we utilized tafazzin-knockout C2C12 mouse myoblasts (TAZ-KO) and cardiac and skeletal muscle tissue from tafazzin-knockout mice to identify an upstream mechanism underlying impaired PDH activity in BTHS. This mechanism centers around robust upregulation of pyruvate dehydrogenase kinase 4 (PDK4), resulting from hyperactivation of AMP-activated protein kinase (AMPK) and subsequent transcriptional upregulation by forkhead box protein O1 (FOXO1). Upregulation of PDK4 in tafazzin-deficient cells causes direct phospho-inhibition of PDH activity accompanied by increased glucose uptake and elevated intracellular glucose concentration. Collectively, our findings provide a novel mechanistic framework whereby impaired tafazzin function ultimately results in robust PDK4 upregulation, leading to impaired PDH activity and likely linked to dysregulated metabolic substrate utilization. This mechanism may underlie previously reported findings of BTHS-associated metabolic dysregulation.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
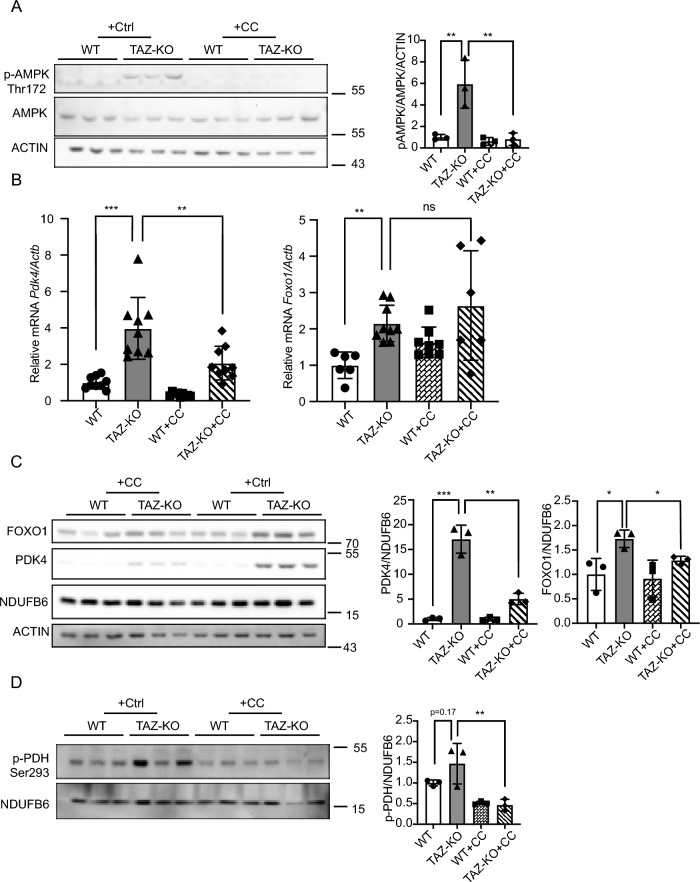
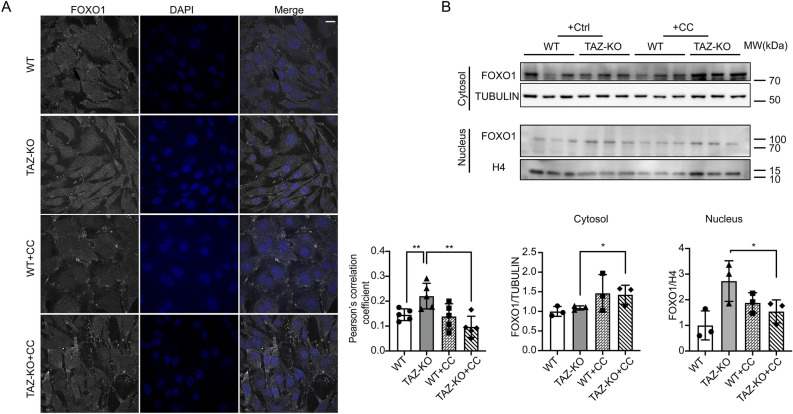
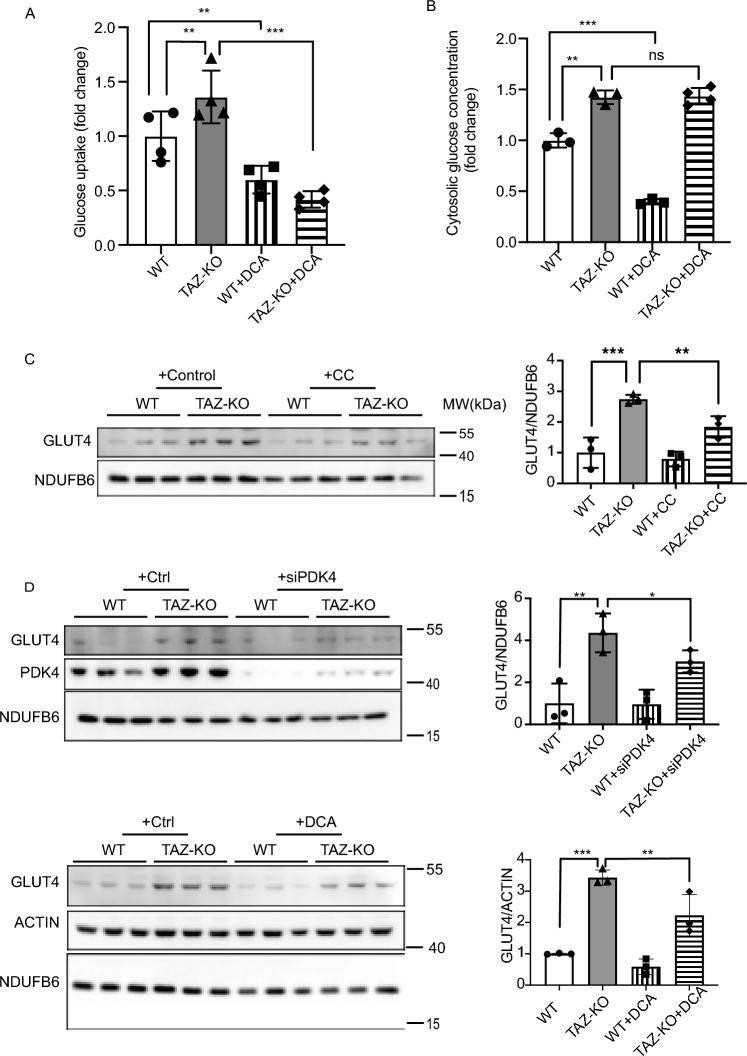
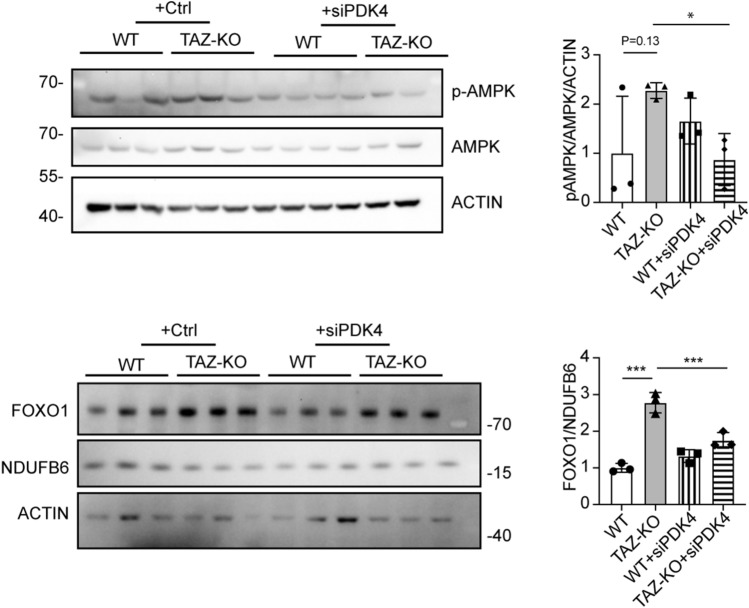
Update of
-
Upregulation of the AMPK-FOXO1-PDK4 pathway is a primary mechanism of pyruvate dehydrogenase activity reduction and leads to increased glucose uptake in tafazzin-deficient cells.bioRxiv [Preprint]. 2024 Feb 4:2024.02.03.578755. doi: 10.1101/2024.02.03.578755. bioRxiv. 2024. Update in: Sci Rep. 2024 May 20;14(1):11497. doi: 10.1038/s41598-024-62262-1. PMID: 38352304 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
