

HATCHet2: clone- and haplotype-specific copy number inference from bulk tumor sequencing data
- PMID: 38773520
- PMCID: PMC11110434
- DOI: 10.1186/s13059-024-03267-x
HATCHet2: clone- and haplotype-specific copy number inference from bulk tumor sequencing data
Abstract
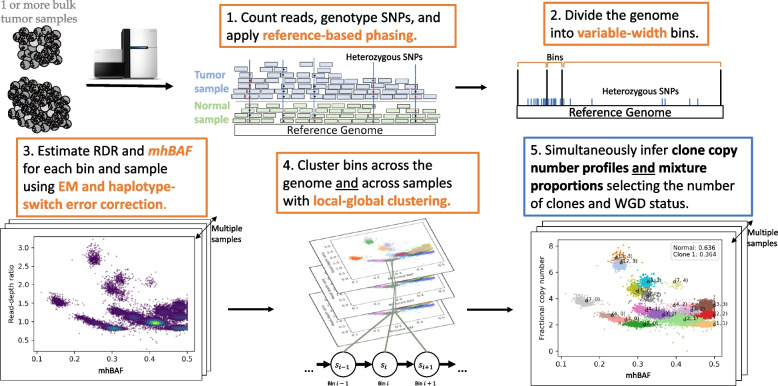
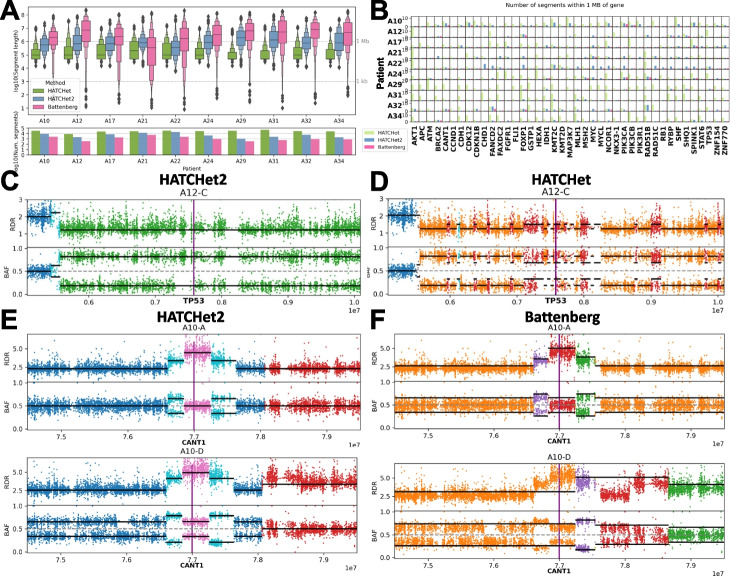
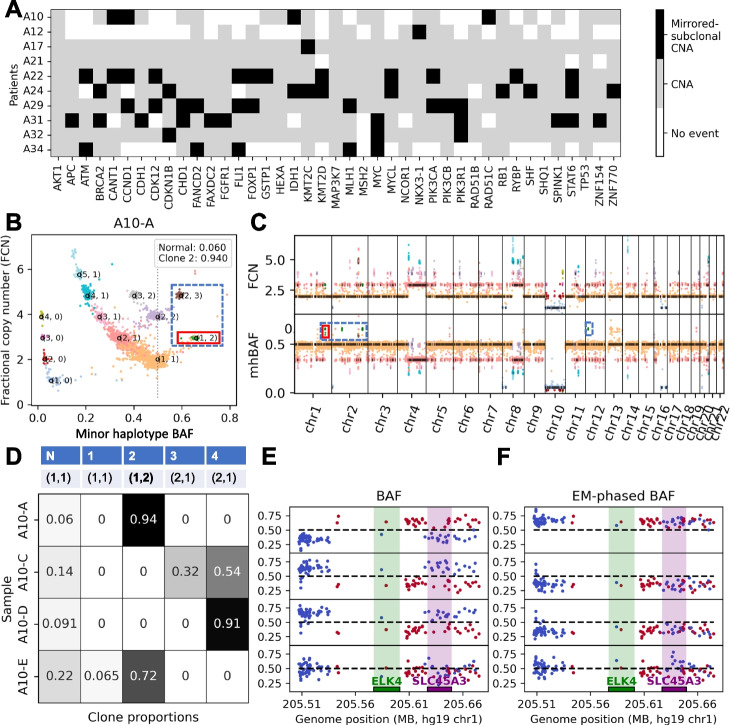
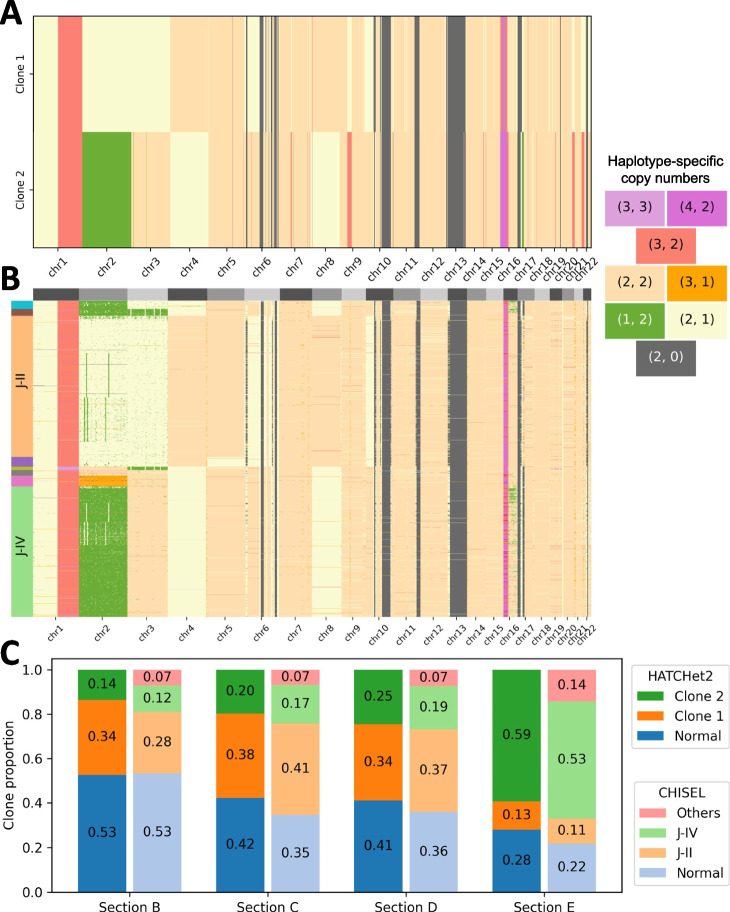
Bulk DNA sequencing of multiple samples from the same tumor is becoming common, yet most methods to infer copy-number aberrations (CNAs) from this data analyze individual samples independently. We introduce HATCHet2, an algorithm to identify haplotype- and clone-specific CNAs simultaneously from multiple bulk samples. HATCHet2 extends the earlier HATCHet method by improving identification of focal CNAs and introducing a novel statistic, the minor haplotype B-allele frequency (mhBAF), that enables identification of mirrored-subclonal CNAs. We demonstrate HATCHet2's improved accuracy using simulations and a single-cell sequencing dataset. HATCHet2 analysis of 10 prostate cancer patients reveals previously unreported mirrored-subclonal CNAs affecting cancer genes.
Keywords: Allele-specific; Cancer; Clone; Copy-number aberrations; DNA sequencing; Genomics; Haplotype; Tumor heterogeneity.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
HATCHet2: clone- and haplotype-specific copy number inference from bulk tumor sequencing data.bioRxiv [Preprint]. 2023 Jul 15:2023.07.13.548855. doi: 10.1101/2023.07.13.548855. bioRxiv. 2023. Update in: Genome Biol. 2024 May 21;25(1):130. doi: 10.1186/s13059-024-03267-x. PMID: 37502835 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
