

Replication stress as a driver of cellular senescence and aging
- PMID: 38777831
- PMCID: PMC11111458
- DOI: 10.1038/s42003-024-06263-w
Replication stress as a driver of cellular senescence and aging
Abstract
Replication stress refers to slowing or stalling of replication fork progression during DNA synthesis that disrupts faithful copying of the genome. While long considered a nexus for DNA damage, the role of replication stress in aging is under-appreciated. The consequential role of replication stress in promotion of organismal aging phenotypes is evidenced by an extensive list of hereditary accelerated aging disorders marked by molecular defects in factors that promote replication fork progression and operate uniquely in the replication stress response. Additionally, recent studies have revealed cellular pathways and phenotypes elicited by replication stress that align with designated hallmarks of aging. Here we review recent advances demonstrating the role of replication stress as an ultimate driver of cellular senescence and aging. We discuss clinical implications of the intriguing links between cellular senescence and aging including application of senotherapeutic approaches in the context of replication stress.
© 2024. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
The authors declare no competing interests.
Figures
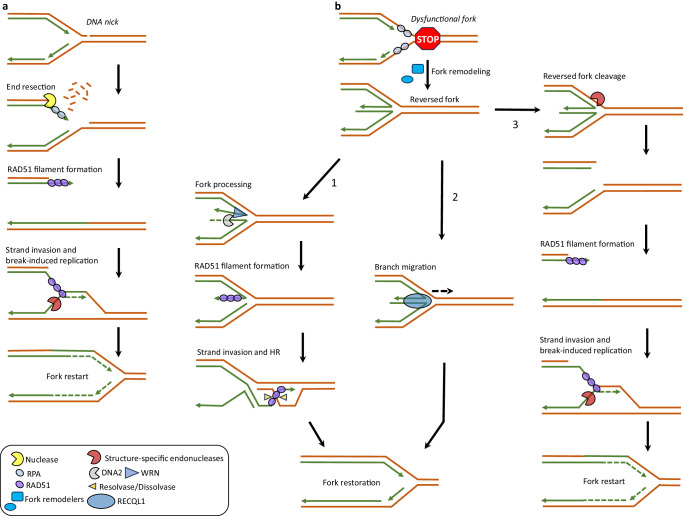

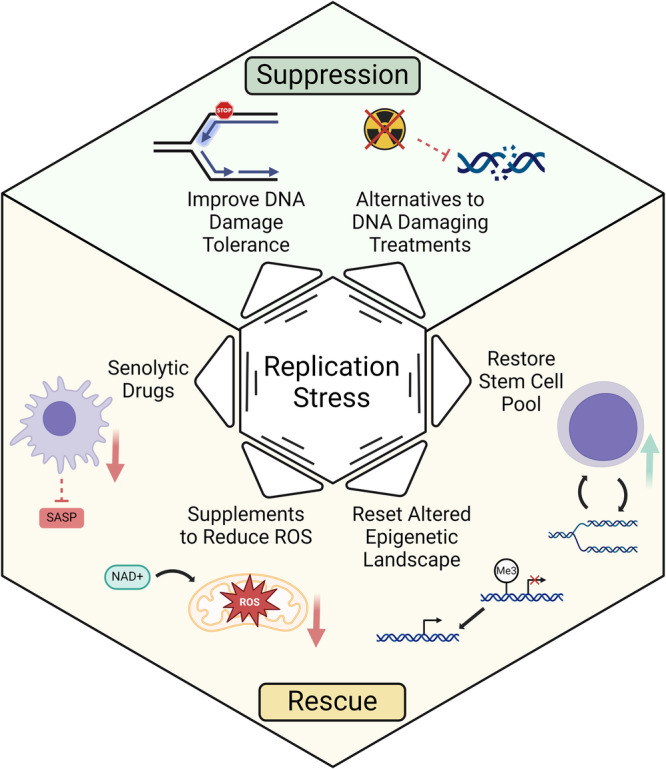
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
