

Natural proteome diversity links aneuploidy tolerance to protein turnover
- PMID: 38778096
- PMCID: PMC11153158
- DOI: 10.1038/s41586-024-07442-9
Natural proteome diversity links aneuploidy tolerance to protein turnover
Abstract
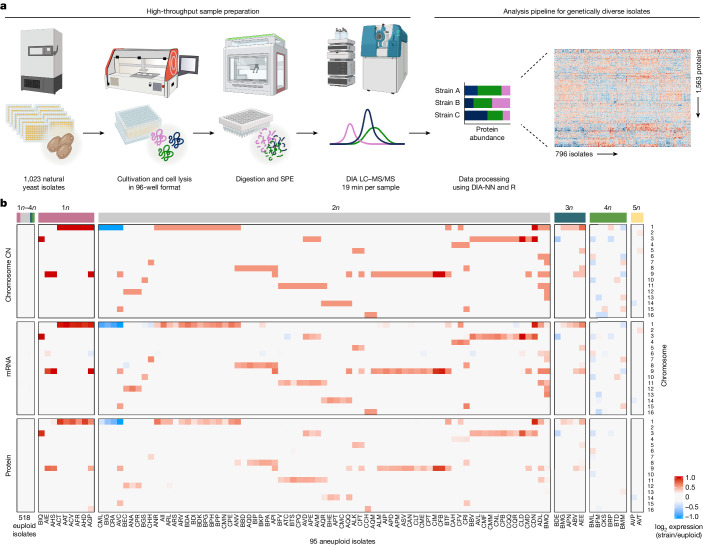
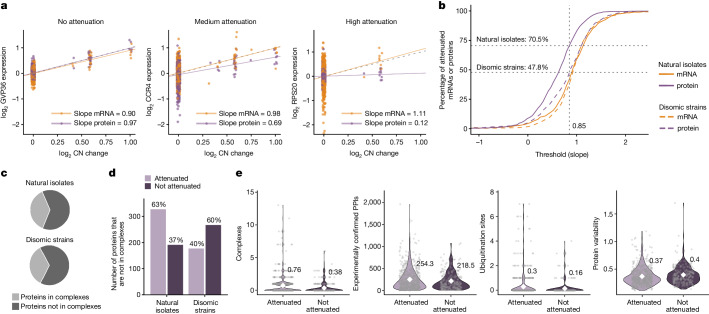
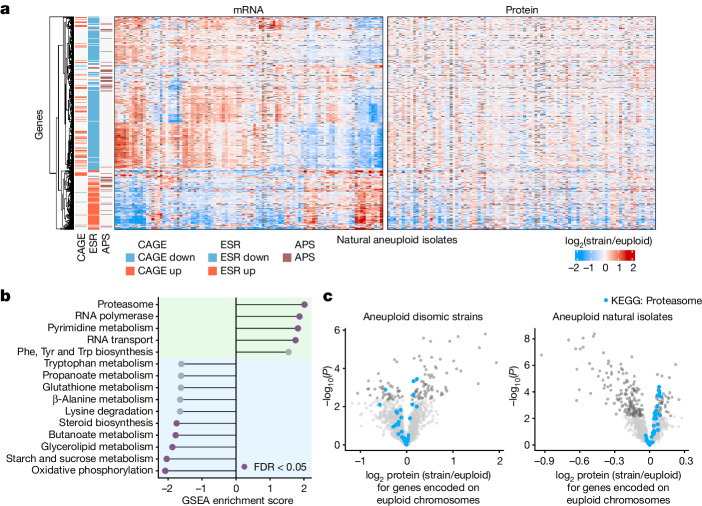
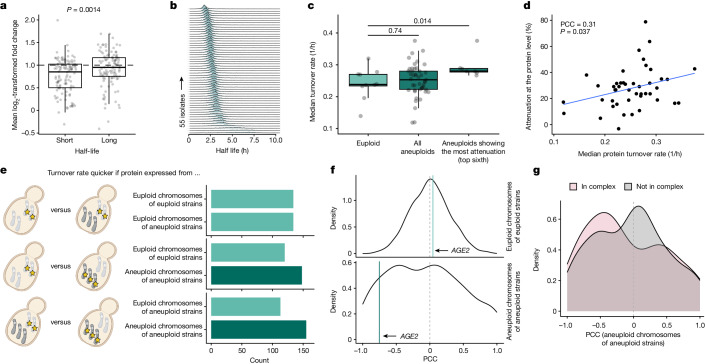
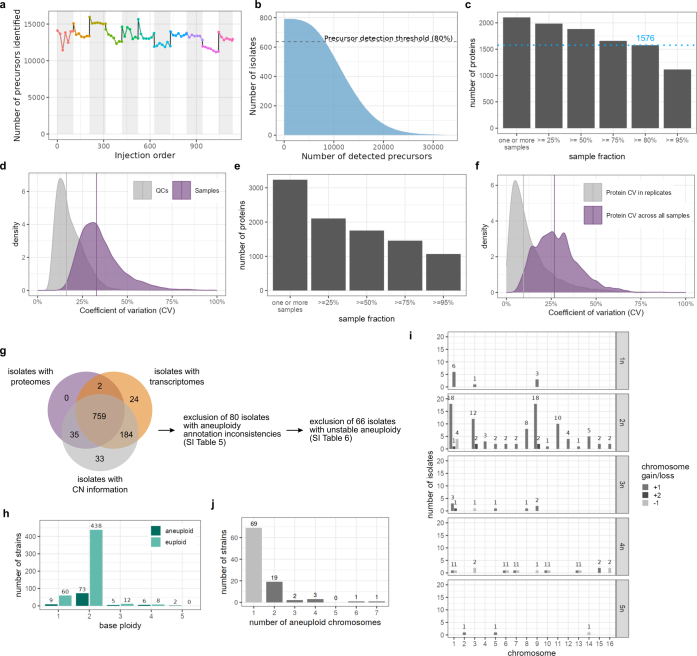
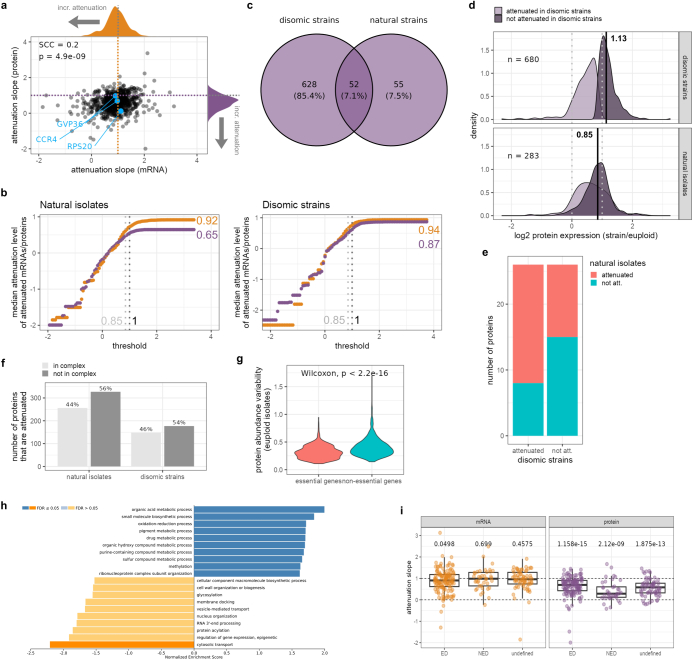
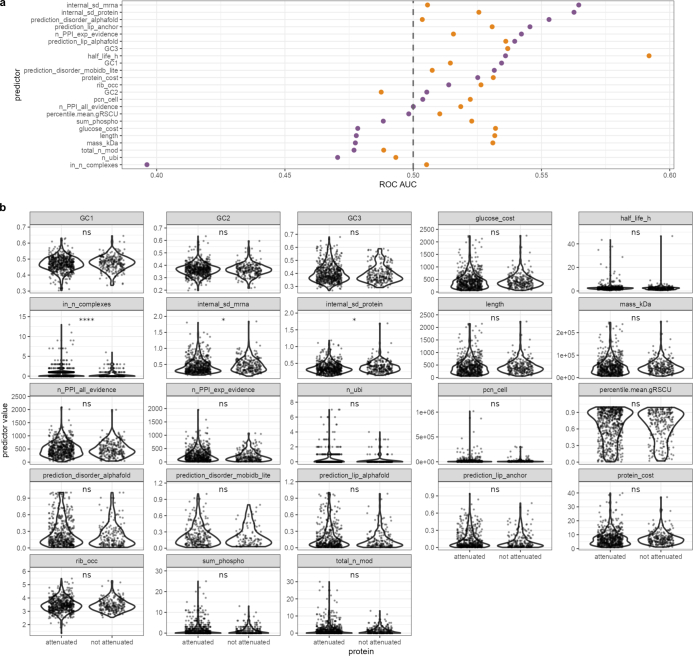
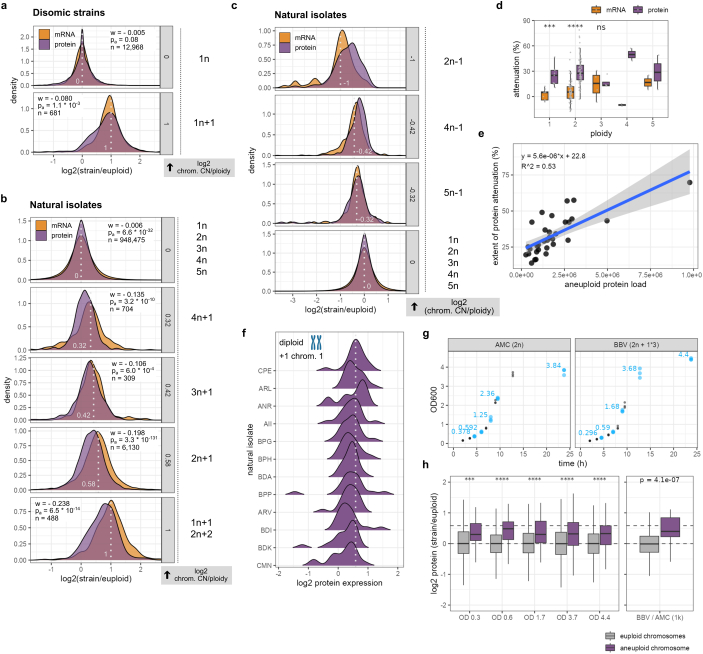
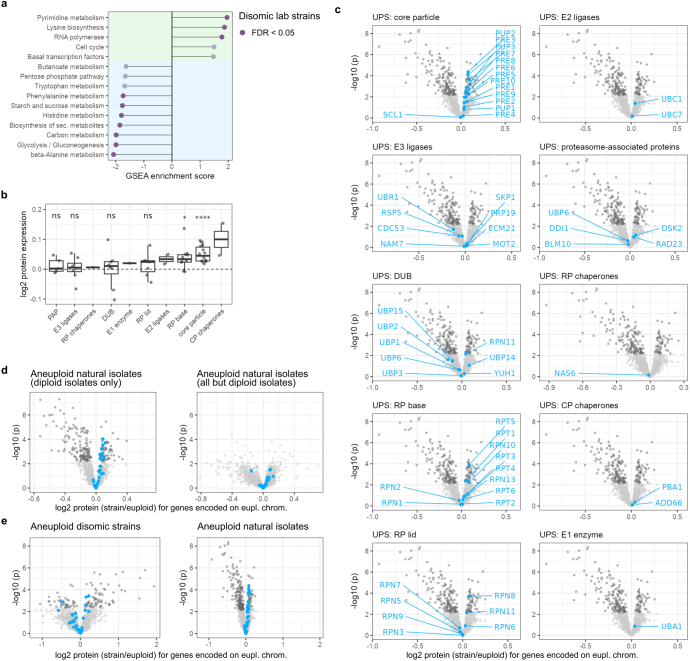
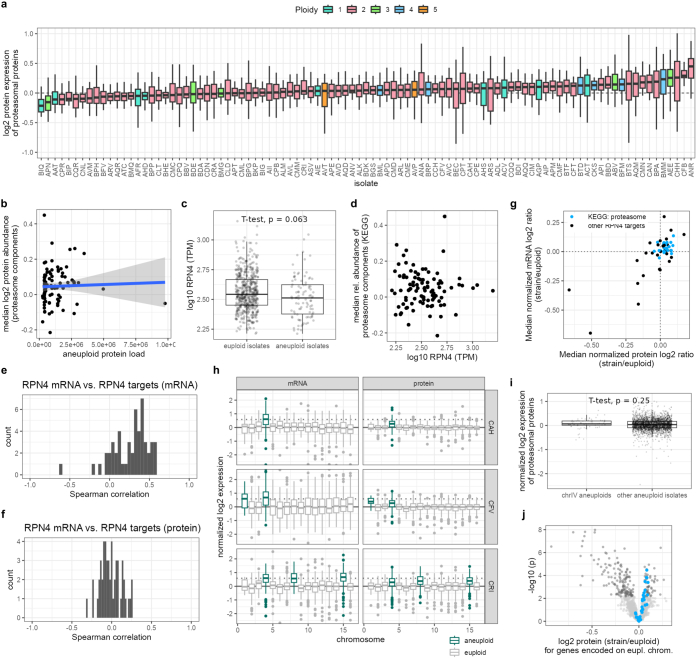
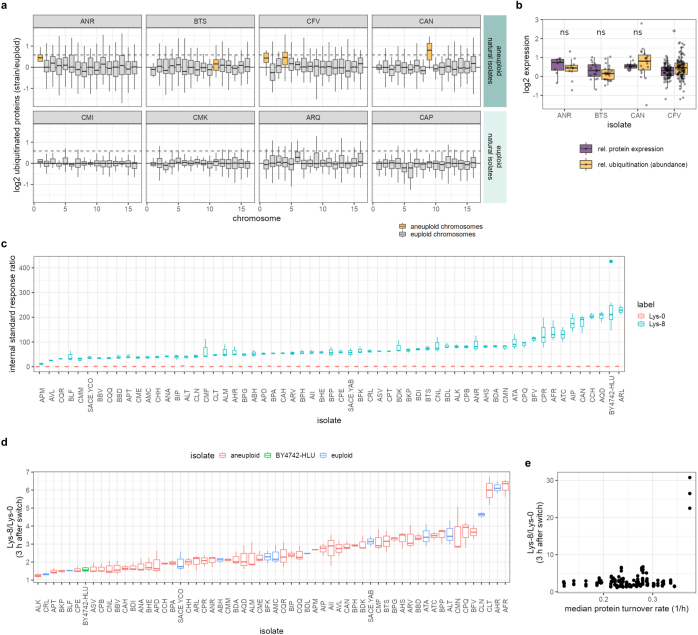
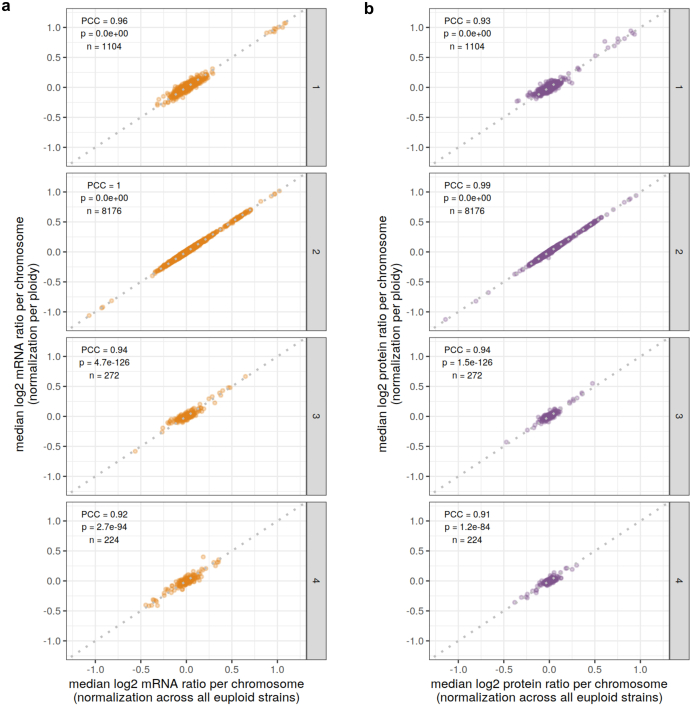
Accessing the natural genetic diversity of species unveils hidden genetic traits, clarifies gene functions and allows the generalizability of laboratory findings to be assessed. One notable discovery made in natural isolates of Saccharomyces cerevisiae is that aneuploidy-an imbalance in chromosome copy numbers-is frequent1,2 (around 20%), which seems to contradict the substantial fitness costs and transient nature of aneuploidy when it is engineered in the laboratory3-5. Here we generate a proteomic resource and merge it with genomic1 and transcriptomic6 data for 796 euploid and aneuploid natural isolates. We find that natural and lab-generated aneuploids differ specifically at the proteome. In lab-generated aneuploids, some proteins-especially subunits of protein complexes-show reduced expression, but the overall protein levels correspond to the aneuploid gene dosage. By contrast, in natural isolates, more than 70% of proteins encoded on aneuploid chromosomes are dosage compensated, and average protein levels are shifted towards the euploid state chromosome-wide. At the molecular level, we detect an induction of structural components of the proteasome, increased levels of ubiquitination, and reveal an interdependency of protein turnover rates and attenuation. Our study thus highlights the role of protein turnover in mediating aneuploidy tolerance, and shows the utility of exploiting the natural diversity of species to attain generalizable molecular insights into complex biological processes.
© 2024. The Author(s).
Conflict of interest statement
M. Steger was an employee of Evotec München. V.D. holds share options of NEOsphere biotechnologies. M.R. is a founder and shareholder of Eliptica. M.M. is a consultant and shareholder of Eliptica. The remaining authors declare no competing interests.
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
