

Mapping medically relevant RNA isoform diversity in the aged human frontal cortex with deep long-read RNA-seq
- PMID: 38778214
- PMCID: PMC11863200
- DOI: 10.1038/s41587-024-02245-9
Mapping medically relevant RNA isoform diversity in the aged human frontal cortex with deep long-read RNA-seq
Abstract
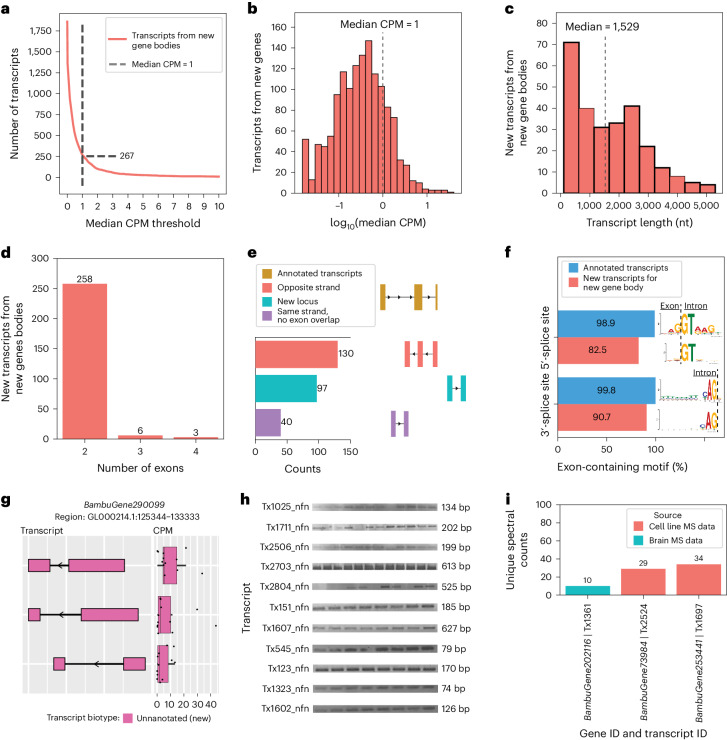
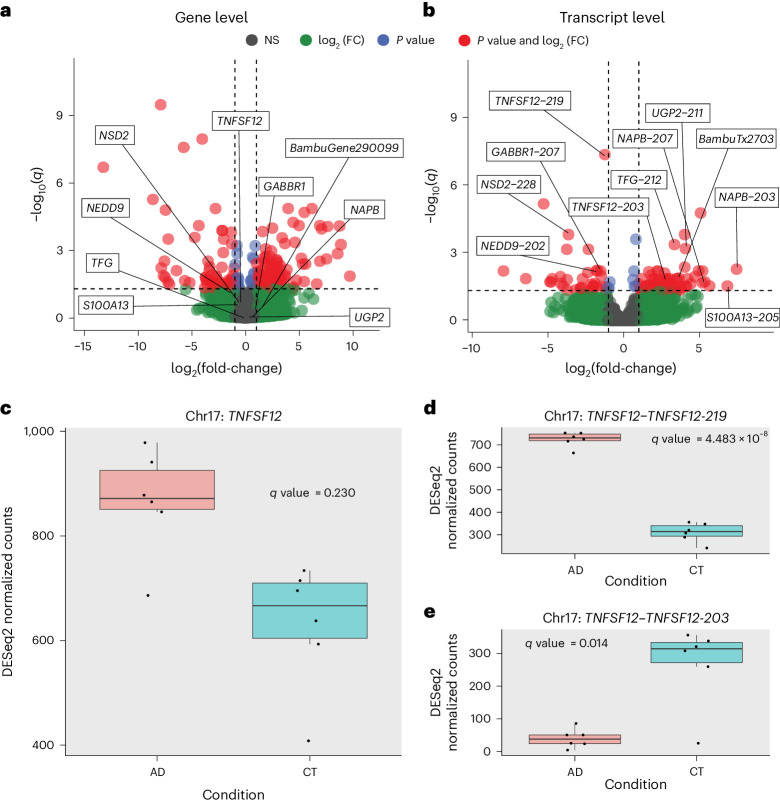
Determining whether the RNA isoforms from medically relevant genes have distinct functions could facilitate direct targeting of RNA isoforms for disease treatment. Here, as a step toward this goal for neurological diseases, we sequenced 12 postmortem, aged human frontal cortices (6 Alzheimer disease cases and 6 controls; 50% female) using one Oxford Nanopore PromethION flow cell per sample. We identified 1,917 medically relevant genes expressing multiple isoforms in the frontal cortex where 1,018 had multiple isoforms with different protein-coding sequences. Of these 1,018 genes, 57 are implicated in brain-related diseases including major depression, schizophrenia, Parkinson's disease and Alzheimer disease. Our study also uncovered 53 new RNA isoforms in medically relevant genes, including several where the new isoform was one of the most highly expressed for that gene. We also reported on five mitochondrially encoded, spliced RNA isoforms. We found 99 differentially expressed RNA isoforms between cases with Alzheimer disease and controls.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
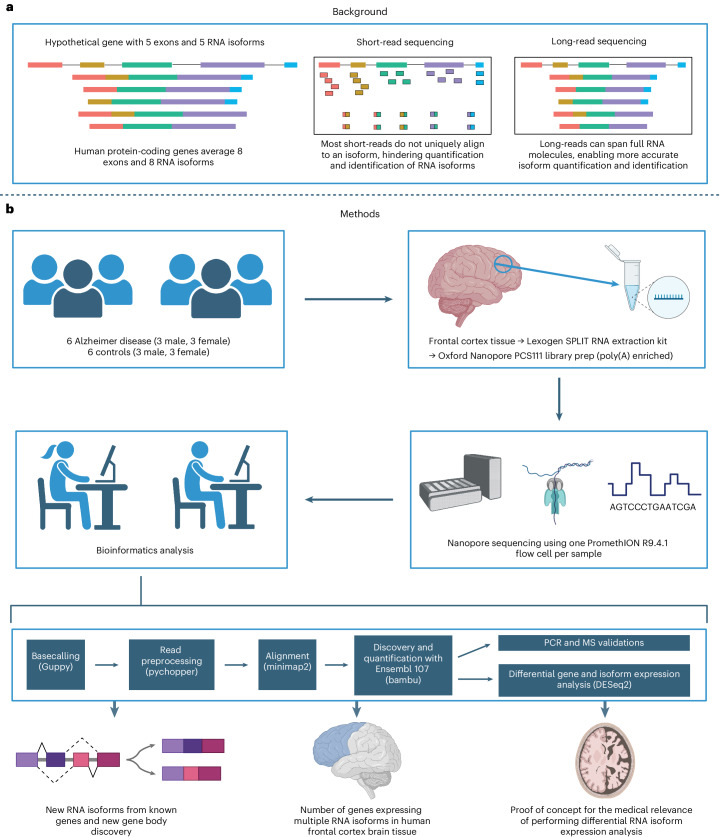













Update of
-
Using deep long-read RNAseq in Alzheimer's disease brain to assess medical relevance of RNA isoform diversity.bioRxiv [Preprint]. 2023 Dec 11:2023.08.06.552162. doi: 10.1101/2023.08.06.552162. bioRxiv. 2023. Update in: Nat Biotechnol. 2025 Apr;43(4):635-646. doi: 10.1038/s41587-024-02245-9. PMID: 37609156 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
- P30 AG072946/AG/NIA NIH HHS/United States
- 2019 -AARG- 644082/ALZ/Alzheimer's Association/United States
- Z01 AG000365/ImNIH/Intramural NIH HHS/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- T32 AG078110/AG/NIA NIH HHS/United States
- RSGTMT17/Pharmaceutical Research and Manufacturers of America Foundation (PhRMA Foundation)
- R01 AG068331/AG/NIA NIH HHS/United States
- 5R50CA243890/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- RF1 AG082339/AG/NIA NIH HHS/United States
- R35 GM138636/GM/NIGMS NIH HHS/United States
- R35GM138636/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- R01AG068331/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
- P30 AG010161/AG/NIA NIH HHS/United States
- R50 CA243890/CA/NCI NIH HHS/United States
- A2020161S/BrightFocus Foundation (BrightFocus)
- R01 AG015819/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
