

Unprecedented genetic variability of PFam54 paralogs among Eurasian Lyme borreliosis-causing spirochetes
- PMID: 38779535
- PMCID: PMC11109050
- DOI: 10.1002/ece3.11397
Unprecedented genetic variability of PFam54 paralogs among Eurasian Lyme borreliosis-causing spirochetes
Abstract
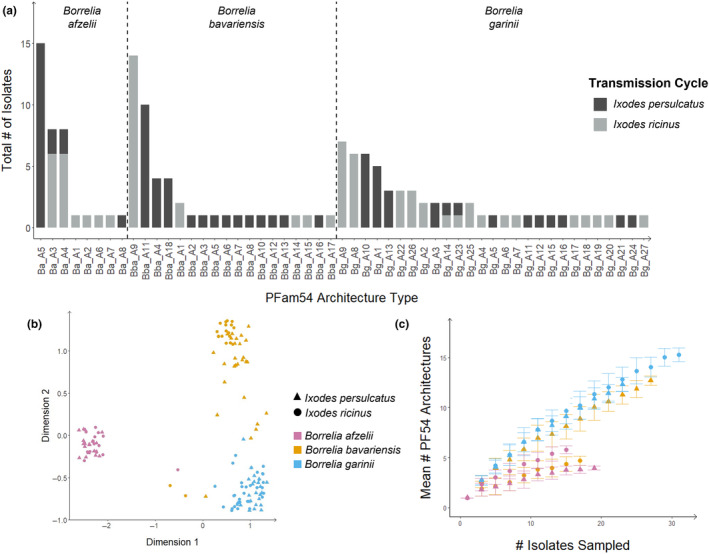
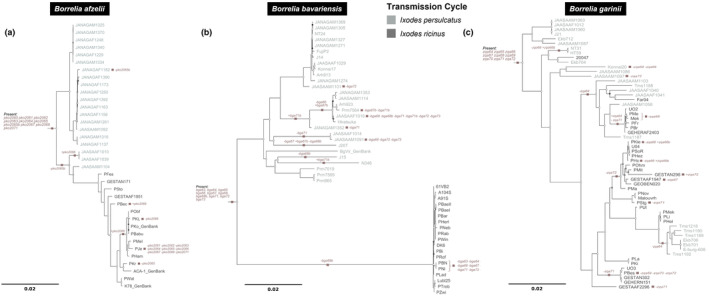
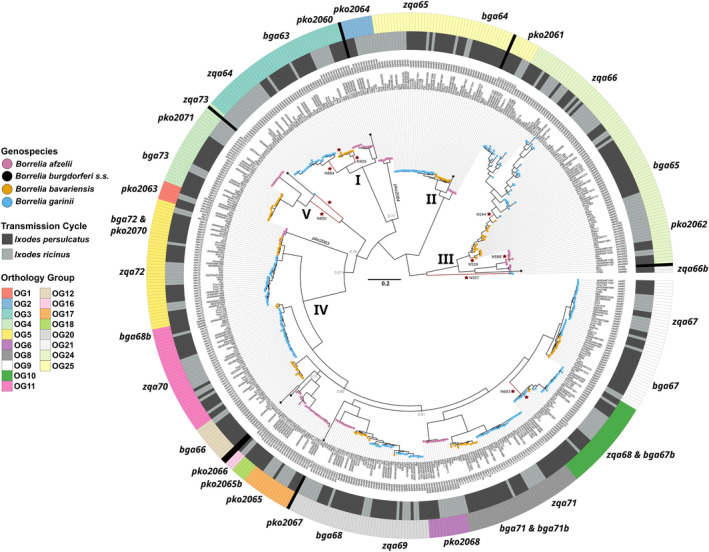
Lyme borreliosis (LB) is the most common vector-borne disease in the Northern Hemisphere caused by spirochetes belonging to the Borrelia burgdorferi sensu lato (Bbsl) complex. Borrelia spirochetes circulate in obligatory transmission cycles between tick vectors and different vertebrate hosts. To successfully complete this complex transmission cycle, Bbsl encodes for an arsenal of proteins including the PFam54 protein family with known, or proposed, influences to reservoir host and/or vector adaptation. Even so, only fragmentary information is available regarding the naturally occurring level of variation in the PFam54 gene array especially in relation to Eurasian-distributed species. Utilizing whole genome data from isolates (n = 141) originated from three major LB-causing Borrelia species across Eurasia (B. afzelii, B. bavariensis, and B. garinii), we aimed to characterize the diversity of the PFam54 gene array in these isolates to facilitate understanding the evolution of PFam54 paralogs on an intra- and interspecies level. We found an extraordinarily high level of variation in the PFam54 gene array with 39 PFam54 paralogs belonging to 23 orthologous groups including five novel paralogs. Even so, the gene array appears to have remained fairly stable over the evolutionary history of the studied Borrelia species. Interestingly, genes outside Clade IV, which contains genes encoding for proteins associated with Borrelia pathogenesis, more frequently displayed signatures of diversifying selection between clades that differ in hypothesized vector or host species. This could suggest that non-Clade IV paralogs play a more important role in host and/or vector adaptation than previously expected, which would require future lab-based studies to validate.
Keywords: Borrelia burgdorferi sensu lato; Lyme borreliosis; PFam54 gene array; gene evolution; host‐pathogen interactions; spirochetes.
© 2024 The Authors. Ecology and Evolution published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors have no conflicts of interest to state.
Figures
References
-
- Atkinson, J. P. , Du Clos, T. W. , Mold, C. , Kulkarni, H. , Hourcade, D. , & Wu, X. (2019). 21 ‐ the human complement system: Basic concepts and clinical relevance. In Rich R. R., Fleisher T. A., Shearer W. T., Schroeder H. W., Frew A. J., & Weyand C. M. (Eds.), Clinical immunology (5th ed., pp. 299–317). Elsevier.
-
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A. A. , Dvorkin, M. , Kulikov, A. S. , Lesin, V. M. , Nikolenko, S. I. , Pham, S. , Prjibelski, A. D. , Pyshkin, A. V. , Sirotkin, A. V. , Vyahhi, N. , Tesler, G. , Alekseyev, M. A. , & Pevzner, P. A. (2012). SPAdes: A New Genome Assembly Algorithm and Its Applications to Single‐Cell Sequencing. Journal of Computational Biology, 19(5), 455–477. 10.1089/cmb.2012.0021 - DOI - PMC - PubMed
-
- Becker, N. S. , Rollins, R. E. , Nosenko, K. , Paulus, A. , Martin, S. , Krebs, S. , Takano, A. , Sato, K. , Kovalev, S. Y. , Kawabata, H. , Fingerle, V. , & Margos, G. (2020). High conservation combined with high plasticity: Genomics and evolution of Borrelia bavariensis . BMC Genomics, 21, 702. 10.1186/s12864-020-07054-3 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
