

Integrative analysis of transcriptome, DNA methylome, and chromatin accessibility reveals candidate therapeutic targets in hypertrophic cardiomyopathy
- PMID: 38780967
- PMCID: PMC11528543
- DOI: 10.1093/procel/pwae032
Integrative analysis of transcriptome, DNA methylome, and chromatin accessibility reveals candidate therapeutic targets in hypertrophic cardiomyopathy
Abstract
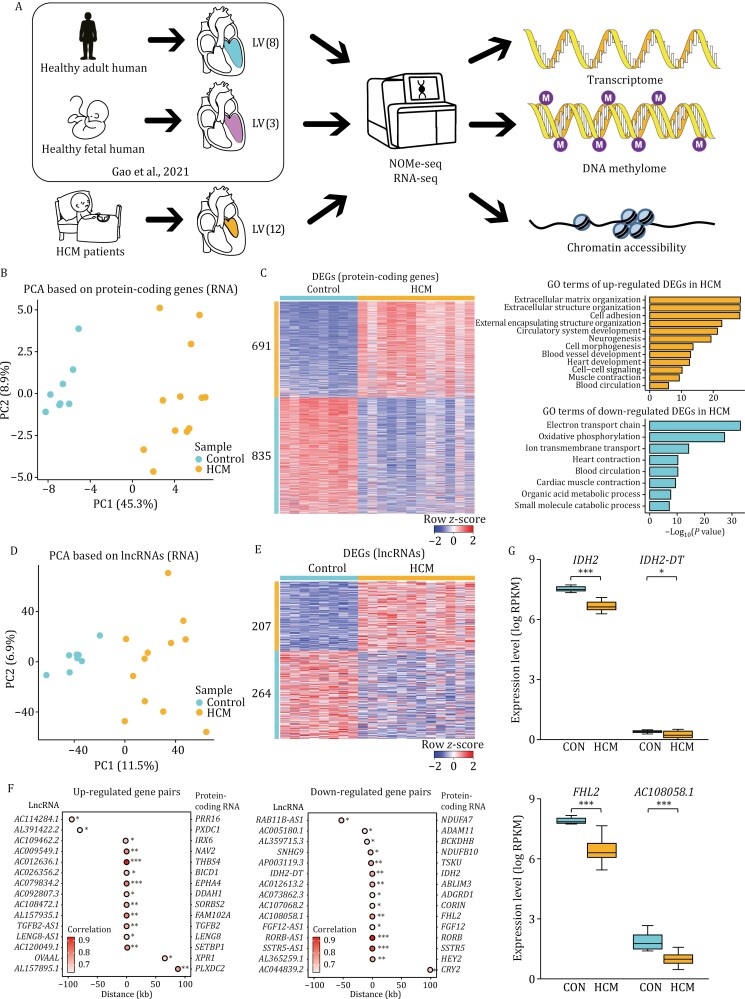
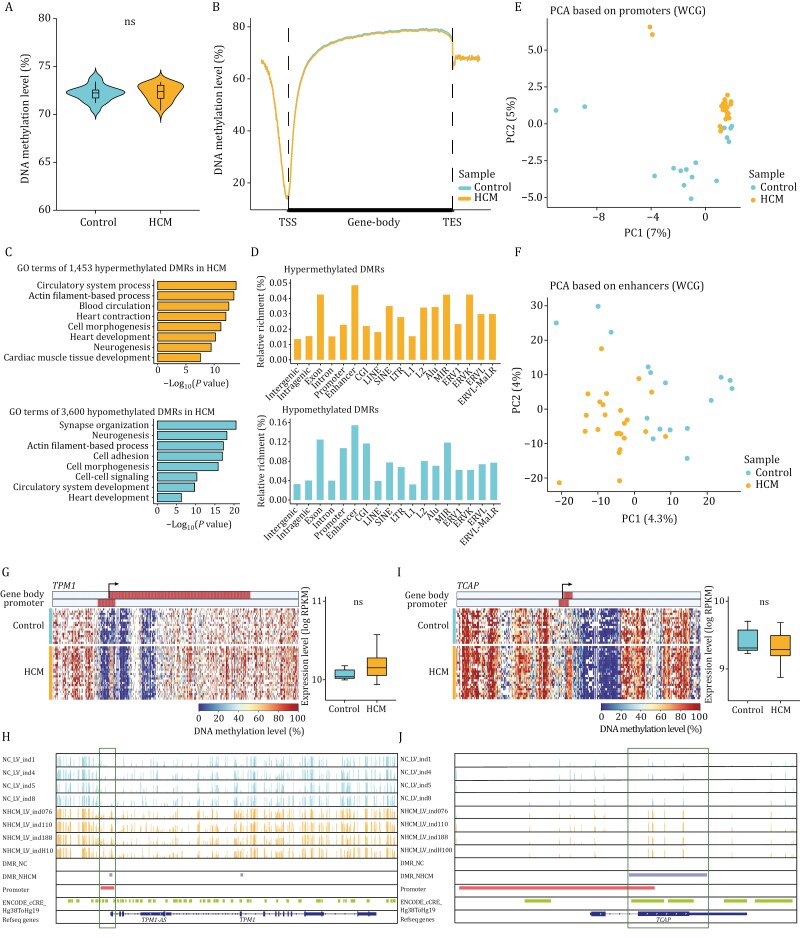
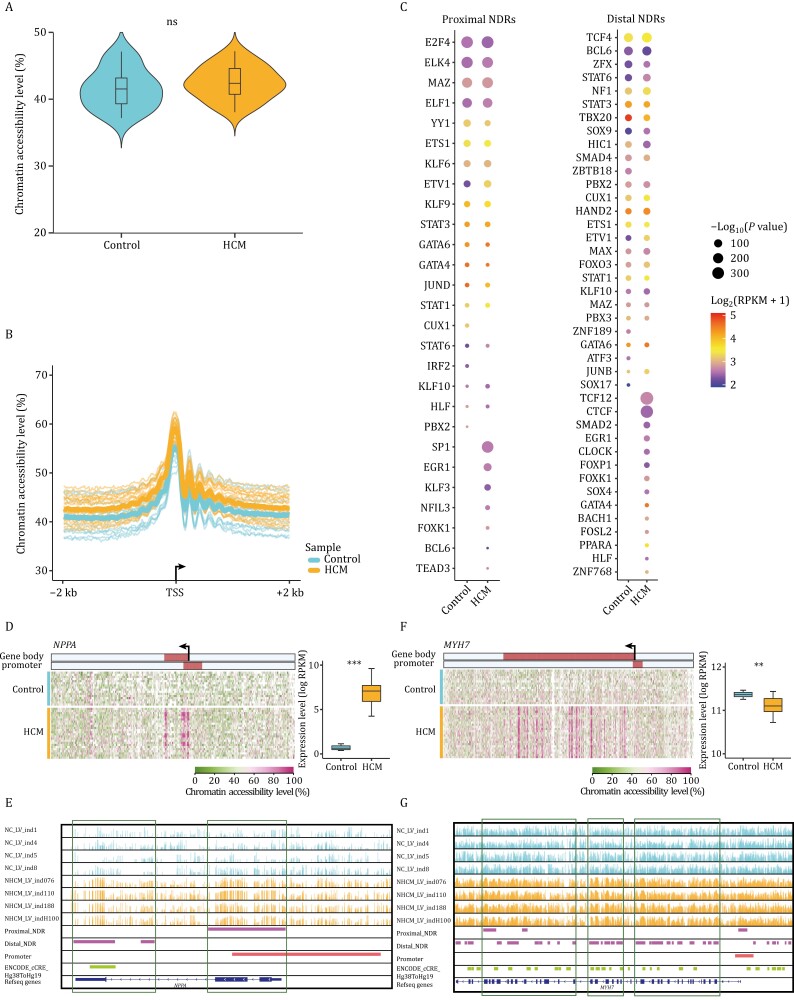
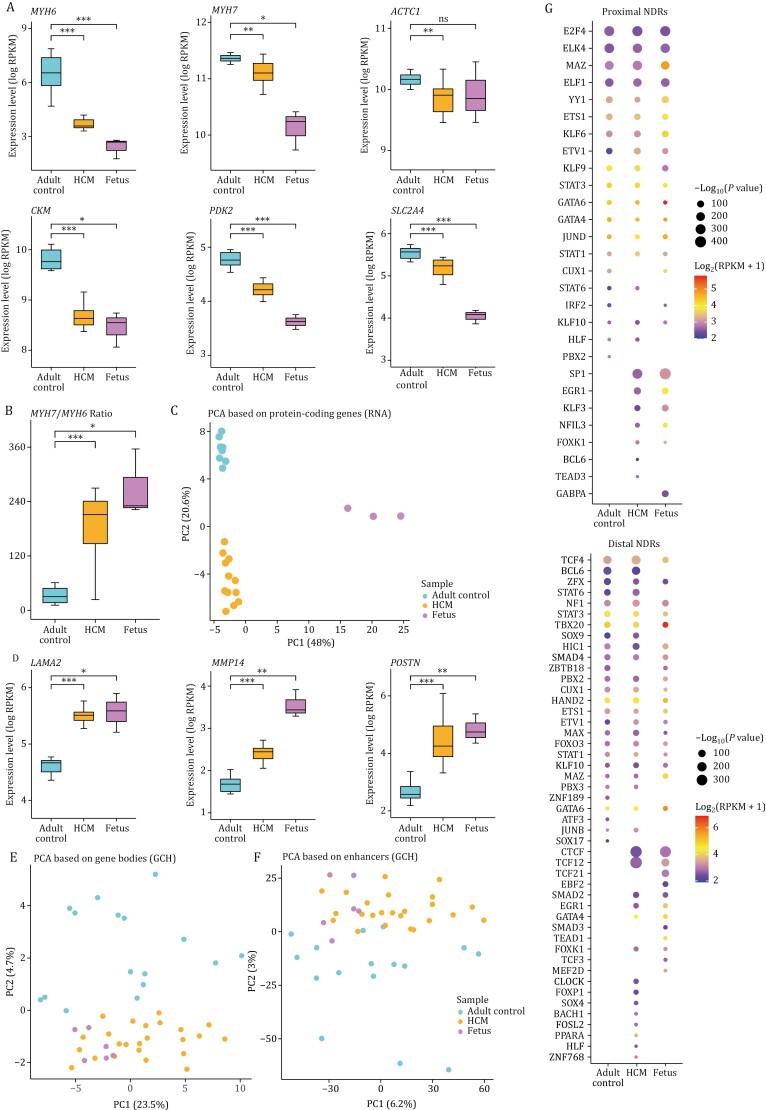
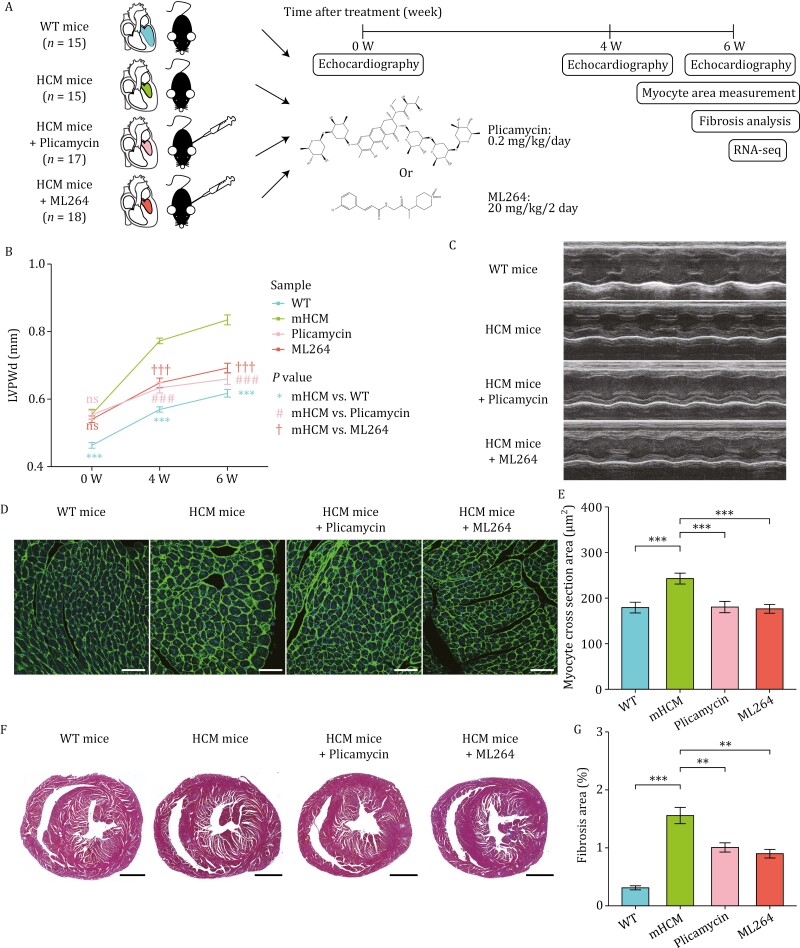
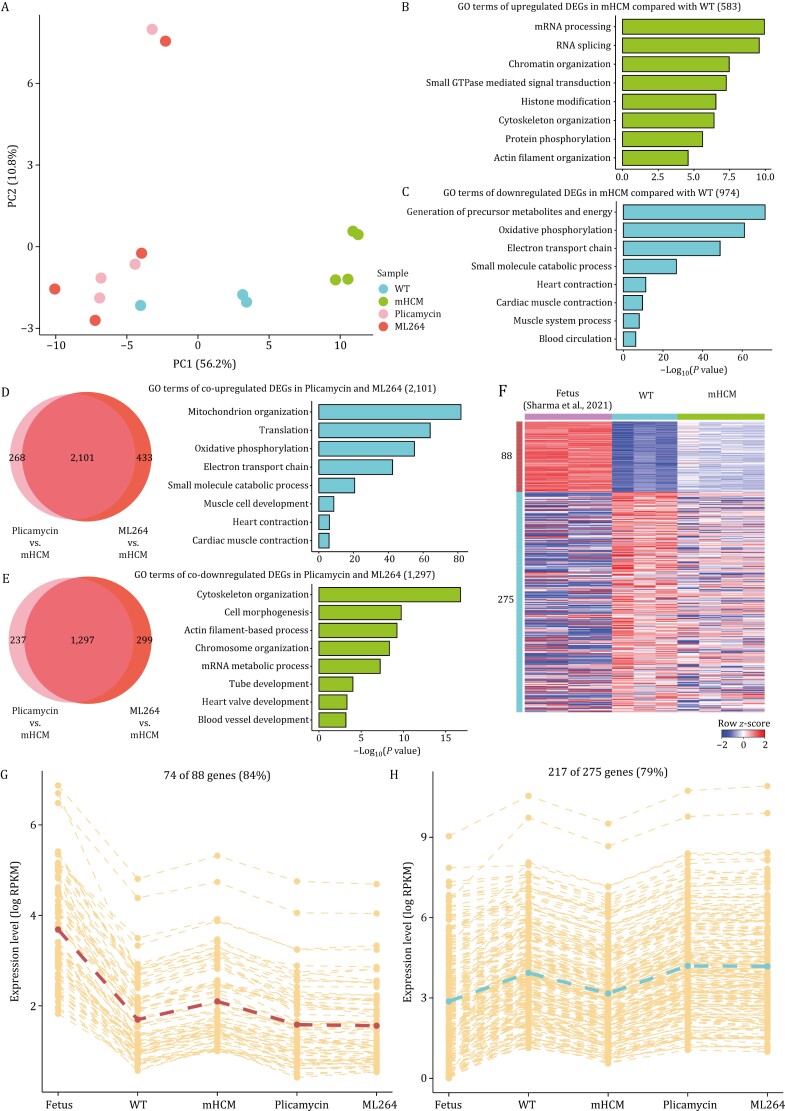
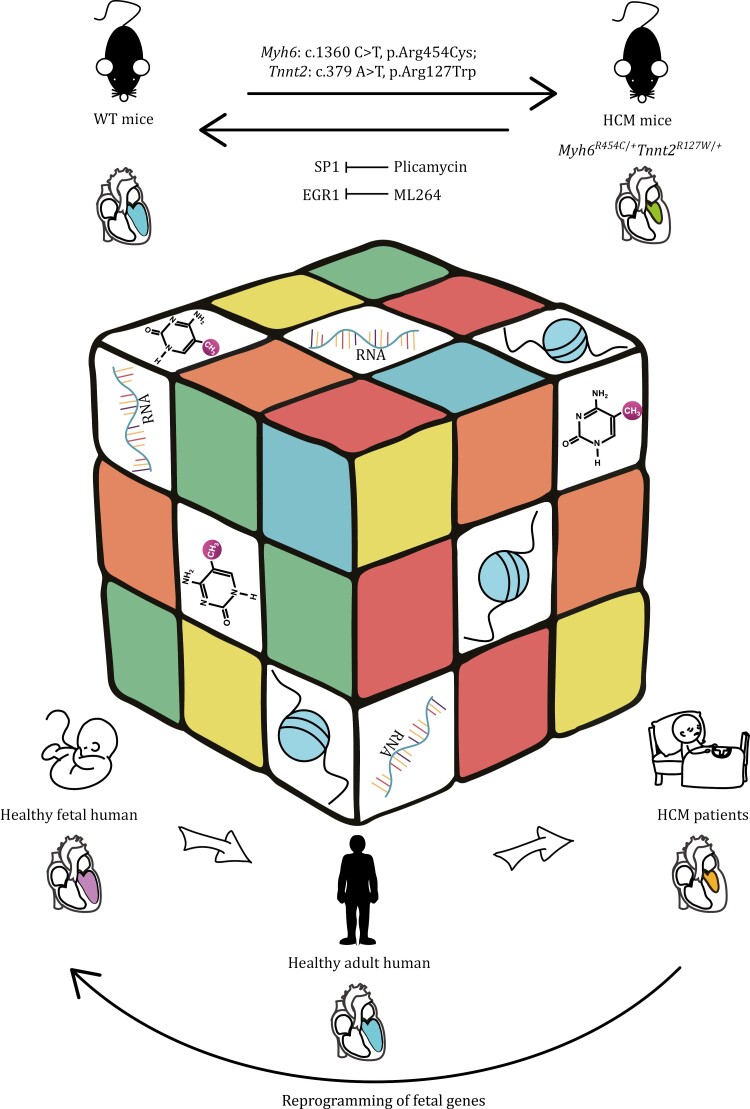
Hypertrophic cardiomyopathy (HCM) is the most common inherited heart disease and is characterized by primary left ventricular hypertrophy usually caused by mutations in sarcomere genes. The mechanism underlying cardiac remodeling in HCM remains incompletely understood. An investigation of HCM through integrative analysis at multi-omics levels will be helpful for treating HCM. DNA methylation and chromatin accessibility, as well as gene expression, were assessed by nucleosome occupancy and methylome sequencing (NOMe-seq) and RNA-seq, respectively, using the cardiac tissues of HCM patients. Compared with those of the controls, the transcriptome, DNA methylome, and chromatin accessibility of the HCM myocardium showed multifaceted differences. At the transcriptome level, HCM hearts returned to the fetal gene program through decreased sarcomeric and metabolic gene expression and increased extracellular matrix gene expression. In the DNA methylome, hypermethylated and hypomethylated differentially methylated regions were identified in HCM. At the chromatin accessibility level, HCM hearts showed changes in different genome elements. Several transcription factors, including SP1 and EGR1, exhibited a fetal-like pattern of binding motifs in nucleosome-depleted regions in HCM. In particular, the inhibition of SP1 or EGR1 in an HCM mouse model harboring sarcomere mutations markedly alleviated the HCM phenotype of the mutant mice and reversed fetal gene reprogramming. Overall, this study not only provides a high-precision multi-omics map of HCM heart tissue but also sheds light on the therapeutic strategy by intervening in the fetal gene reprogramming in HCM.
Keywords: DNA methylation; chromatin accessibility; fetal gene reprogramming; hypertrophic cardiomyopathy; multi-omics; therapy.
© The Author(s) 2024. Published by Oxford University Press on behalf of Higher Education Press.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
DNA methylome and transcriptome profiling reveal key electrophysiology and immune dysregulation in hypertrophic cardiomyopathy.Epigenetics. 2023 Dec;18(1):2195307. doi: 10.1080/15592294.2023.2195307. Epigenetics. 2023. PMID: 37005704 Free PMC article.
-
Multi-omics integration identifies key upstream regulators of pathomechanisms in hypertrophic cardiomyopathy due to truncating MYBPC3 mutations.Clin Epigenetics. 2021 Mar 23;13(1):61. doi: 10.1186/s13148-021-01043-3. Clin Epigenetics. 2021. PMID: 33757590 Free PMC article.
-
Differences in molecular phenotype in mouse and human hypertrophic cardiomyopathy.Sci Rep. 2021 Jun 23;11(1):13163. doi: 10.1038/s41598-021-89451-6. Sci Rep. 2021. PMID: 34162896 Free PMC article.
-
Hypertrophic Cardiomyopathy: A Vicious Cycle Triggered by Sarcomere Mutations and Secondary Disease Hits.Antioxid Redox Signal. 2019 Aug 1;31(4):318-358. doi: 10.1089/ars.2017.7236. Epub 2018 Apr 11. Antioxid Redox Signal. 2019. PMID: 29490477 Free PMC article. Review.
-
The molecular genetic basis for hypertrophic cardiomyopathy.J Mol Cell Cardiol. 2001 Apr;33(4):655-70. doi: 10.1006/jmcc.2001.1340. J Mol Cell Cardiol. 2001. PMID: 11273720 Free PMC article. Review.
Cited by
-
Genetic insights into idiopathic pulmonary fibrosis: a multi-omics approach to identify potential therapeutic targets.J Transl Med. 2025 Mar 16;23(1):337. doi: 10.1186/s12967-025-06368-8. J Transl Med. 2025. PMID: 40091050 Free PMC article.
-
Analysis of human brain RNA-seq data reveals combined effects of 4 types of RNA modifications and 18 types of programmed cell death on Alzheimer's disease.J Transl Med. 2025 Apr 3;23(1):396. doi: 10.1186/s12967-025-06324-6. J Transl Med. 2025. PMID: 40181382 Free PMC article.
-
Clinical and Genetic Heterogeneity of HCM: The Possible Role of a Deletion Involving MYH6 and MYH7.Genes (Basel). 2025 Feb 10;16(2):212. doi: 10.3390/genes16020212. Genes (Basel). 2025. PMID: 40004541 Free PMC article.
-
Transcriptomic analysis and machine learning modeling identifies novel biomarkers and genetic characteristics of hypertrophic cardiomyopathy.Front Genet. 2025 Jun 17;16:1596049. doi: 10.3389/fgene.2025.1596049. eCollection 2025. Front Genet. 2025. PMID: 40599543 Free PMC article.
-
Generic and queryable data integration schema for transcriptomics and epigenomics studies.Comput Struct Biotechnol J. 2024 Nov 19;23:4232-4241. doi: 10.1016/j.csbj.2024.11.022. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 39660218 Free PMC article.
References
-
- Azakie A, Fineman JR, He Y.. Myocardial transcription factors are modulated during pathologic cardiac hypertrophy in vivo. J Thorac Cardiovasc Surg 2006a;132:1262–1271. - PubMed
-
- Azakie A, Fineman JR, He Y.. Sp3 inhibits Sp1-mediated activation of the cardiac troponin T promoter and is downregulated during pathological cardiac hypertrophy in vivo. Am J Physiol Heart Circ Physiol 2006b;291:H600–H611. - PubMed
-
- Boon RA, Jae N, Holdt L. et al. Long noncoding RNAs: from clinical genetics to therapeutic targets? J Am Coll Cardiol 2016;67:1214–1226. - PubMed
-
- Brower GL, Gardner JD, Forman MF. et al. The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur J Cardiothorac Surg 2006;30:604–610. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
