

A synthetic lethal dependency on casein kinase 2 in response to replication-perturbing therapeutics in RB1-deficient cancer cells
- PMID: 38781347
- PMCID: PMC11114247
- DOI: 10.1126/sciadv.adj1564
A synthetic lethal dependency on casein kinase 2 in response to replication-perturbing therapeutics in RB1-deficient cancer cells
Abstract
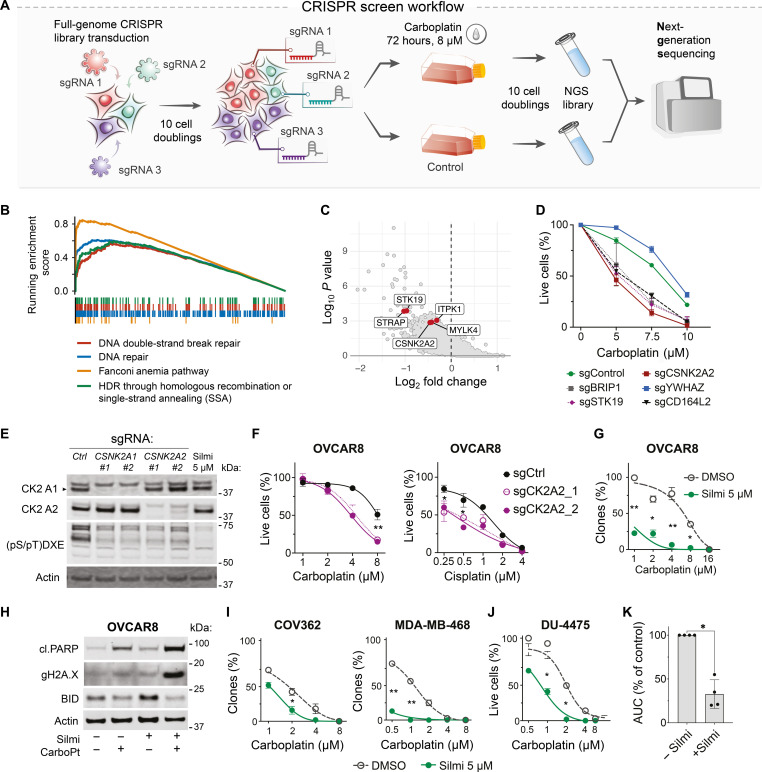
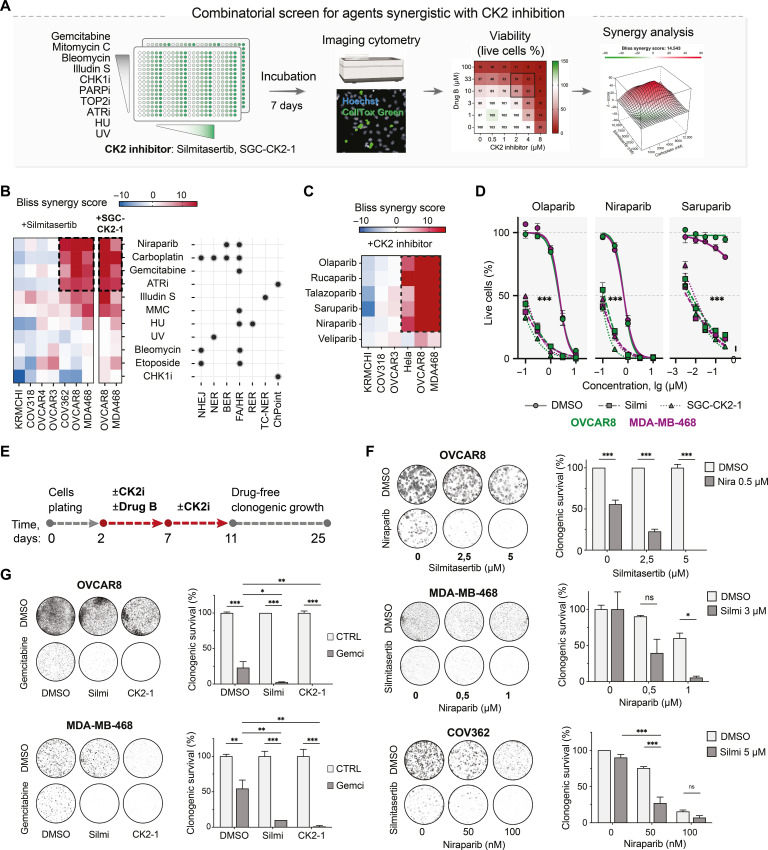
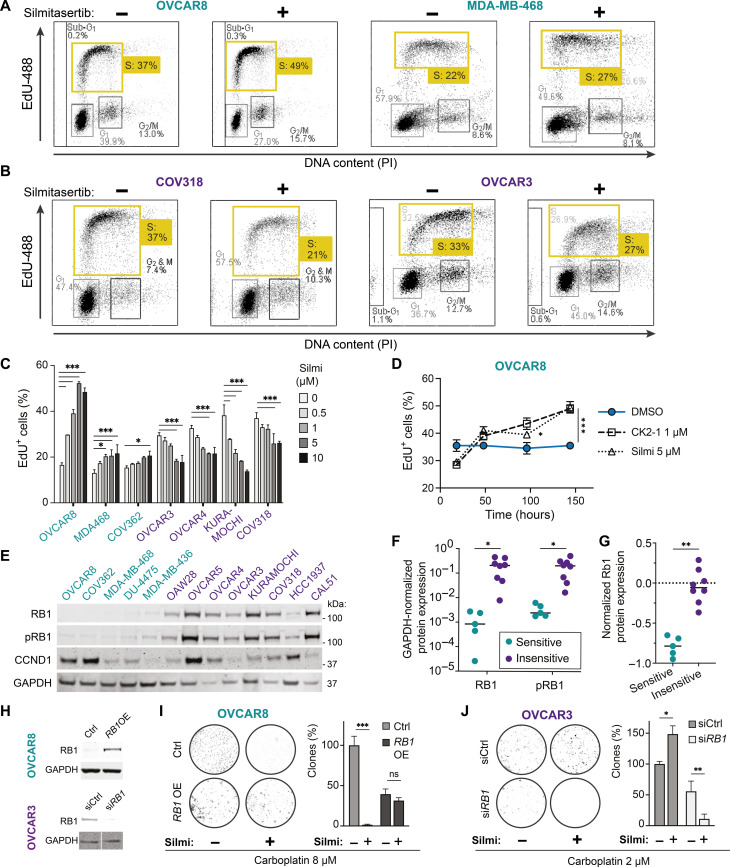
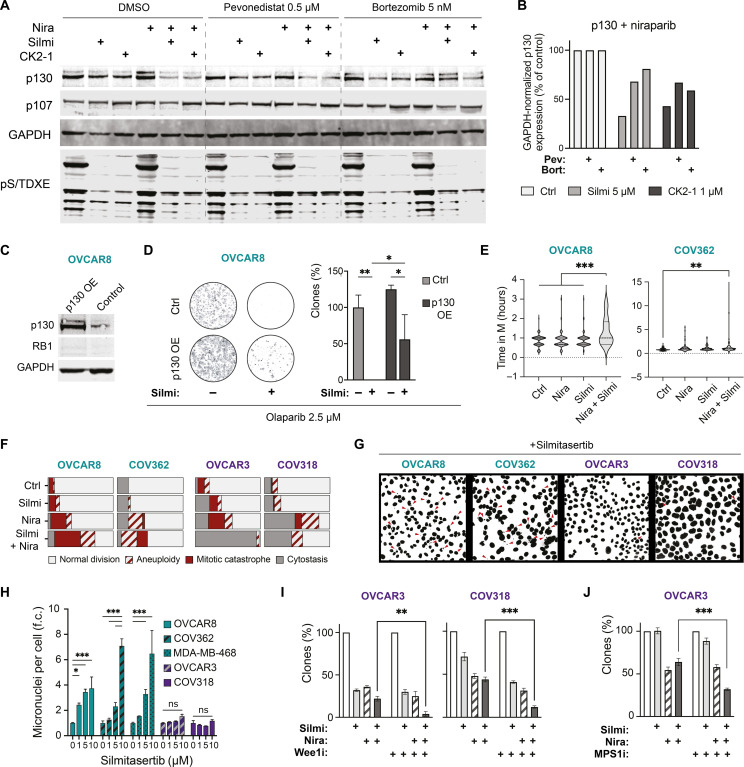
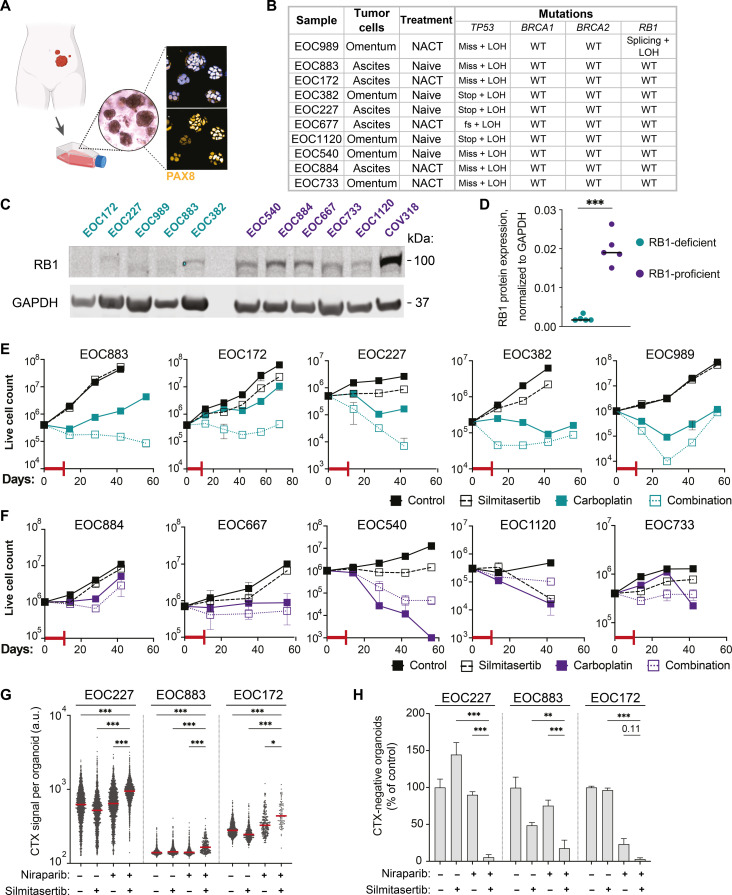
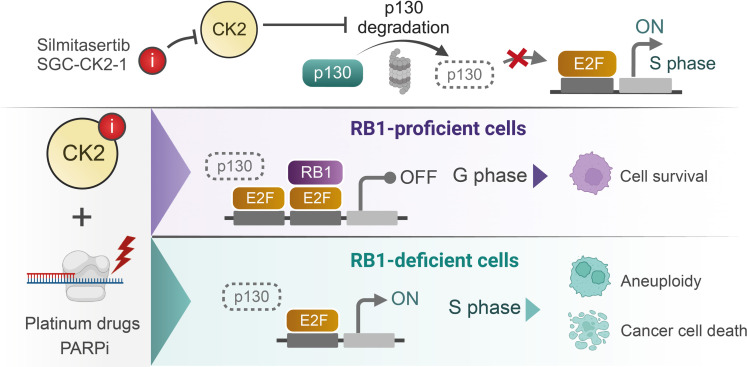
Resistance to therapy commonly develops in patients with high-grade serous ovarian carcinoma (HGSC) and triple-negative breast cancer (TNBC), urging the search for improved therapeutic combinations and their predictive biomarkers. Starting from a CRISPR knockout screen, we identified that loss of RB1 in TNBC or HGSC cells generates a synthetic lethal dependency on casein kinase 2 (CK2) for surviving the treatment with replication-perturbing therapeutics such as carboplatin, gemcitabine, or PARP inhibitors. CK2 inhibition in RB1-deficient cells resulted in the degradation of another RB family cell cycle regulator, p130, which led to S phase accumulation, micronuclei formation, and accelerated PARP inhibition-induced aneuploidy and mitotic cell death. CK2 inhibition was also effective in primary patient-derived cells. It selectively prevented the regrowth of RB1-deficient patient HGSC organoids after treatment with carboplatin or niraparib. As about 25% of HGSCs and 40% of TNBCs have lost RB1 expression, CK2 inhibition is a promising approach to overcome resistance to standard therapeutics in large strata of patients.
Figures
References
-
- Bowtell D. D., Böhm S., Ahmed A. A., Aspuria P.-J., Bast R. C., Beral V., Berek J. S., Birrer M. J., Blagden S., Bookman M. A., Brenton J. D., Chiappinelli K. B., Martins F. C., Coukos G., Drapkin R., Edmondson R., Fotopoulou C., Gabra H., Galon J., Gourley C., Heong V., Huntsman D. G., Iwanicki M., Karlan B. Y., Kaye A., Lengyel E., Levine D. A., Lu K. H., McNeish I. A., Menon U., Narod S. A., Nelson B. H., Nephew K. P., Pharoah P., Powell D. J., Ramos P., Romero I. L., Scott C. L., Sood A. K., Stronach E. A., Balkwill F. R., Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 15, 668–679 (2015). - PMC - PubMed
-
- Bianchini G., De Angelis C., Licata L., Gianni L., Treatment landscape of triple-negative breast cancer —expanded options, evolving needs. Nat. Rev. Clin. Oncol. 19, 91–113 (2021). - PubMed
-
- Tutt A., Robson M., Garber J. E., Domchek S. M., Audeh M. W., Weitzel J. N., Friedlander M., Arun B., Loman N., Schmutzler R. K., Wardley A., Mitchell G., Earl H., Wickens M., Carmichael J., Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 376, 235–244 (2010). - PubMed
-
- Audeh M. W., Audeh M. W., Carmichael J., Penson R. T., Friedlander M., Powell B., Bell-McGuinn K. M., Scott C., Weitzel J. N., Oaknin A., Loman N., Lu K., Schmutzler R. K., Matulonis U., Wickens M., Tutt A., Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 376, 245–251 (2010). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
