

Single-cell genomics and regulatory networks for 388 human brains
- PMID: 38781369
- PMCID: PMC11365579
- DOI: 10.1126/science.adi5199
Single-cell genomics and regulatory networks for 388 human brains
Abstract
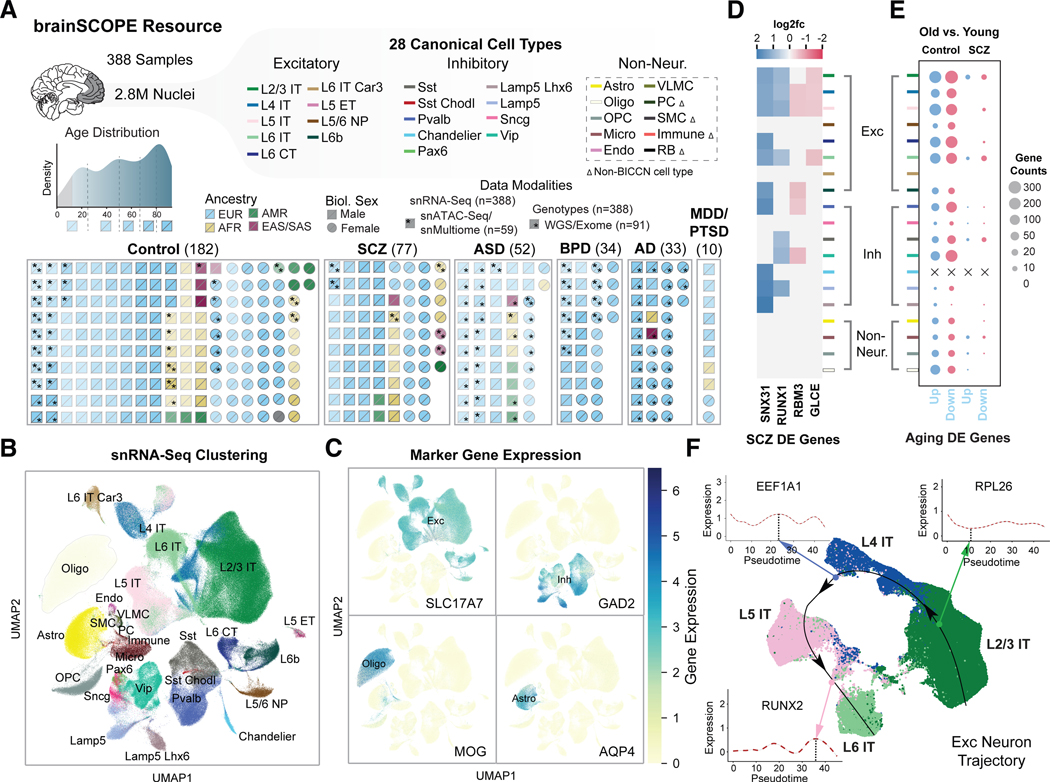
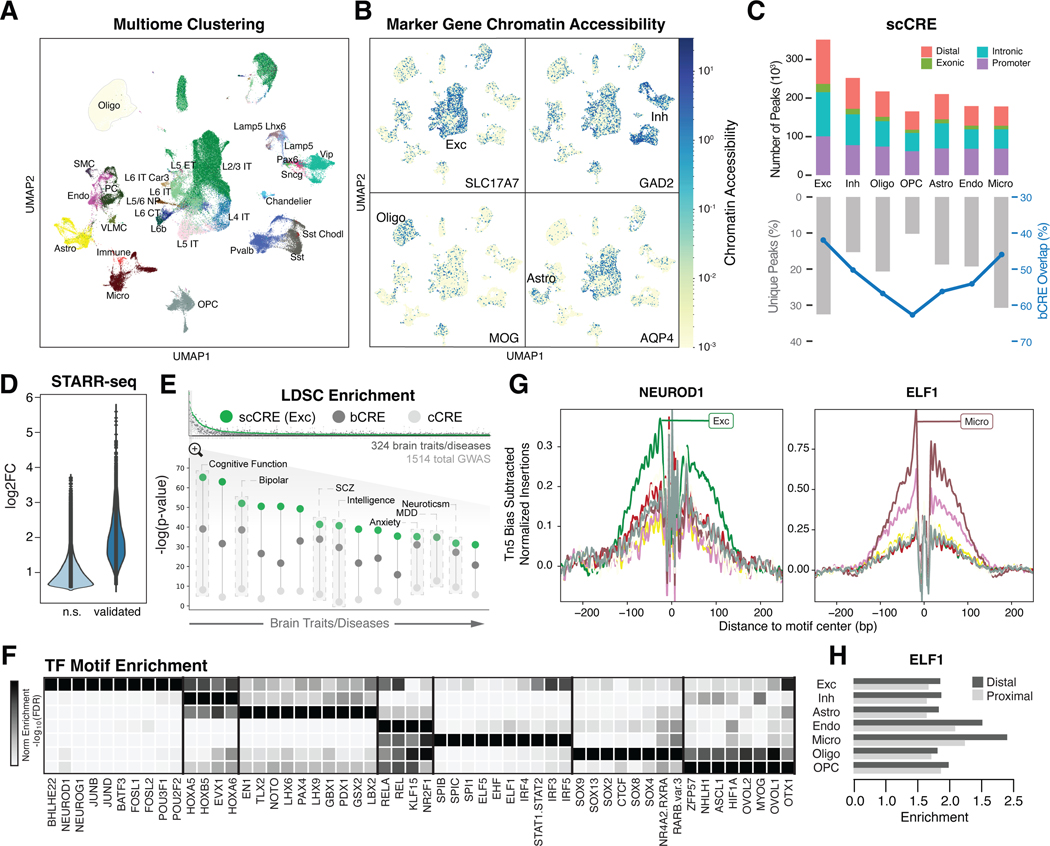
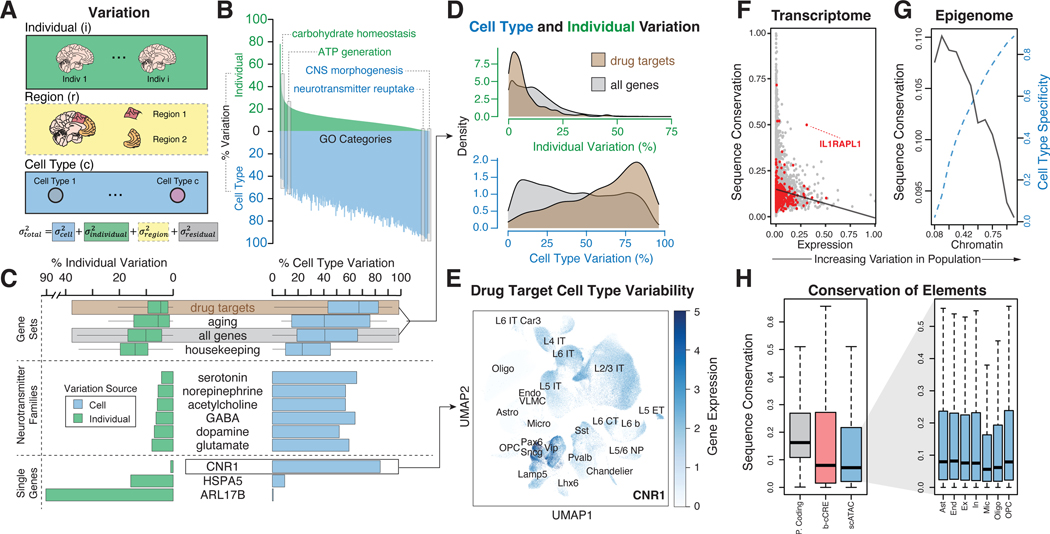
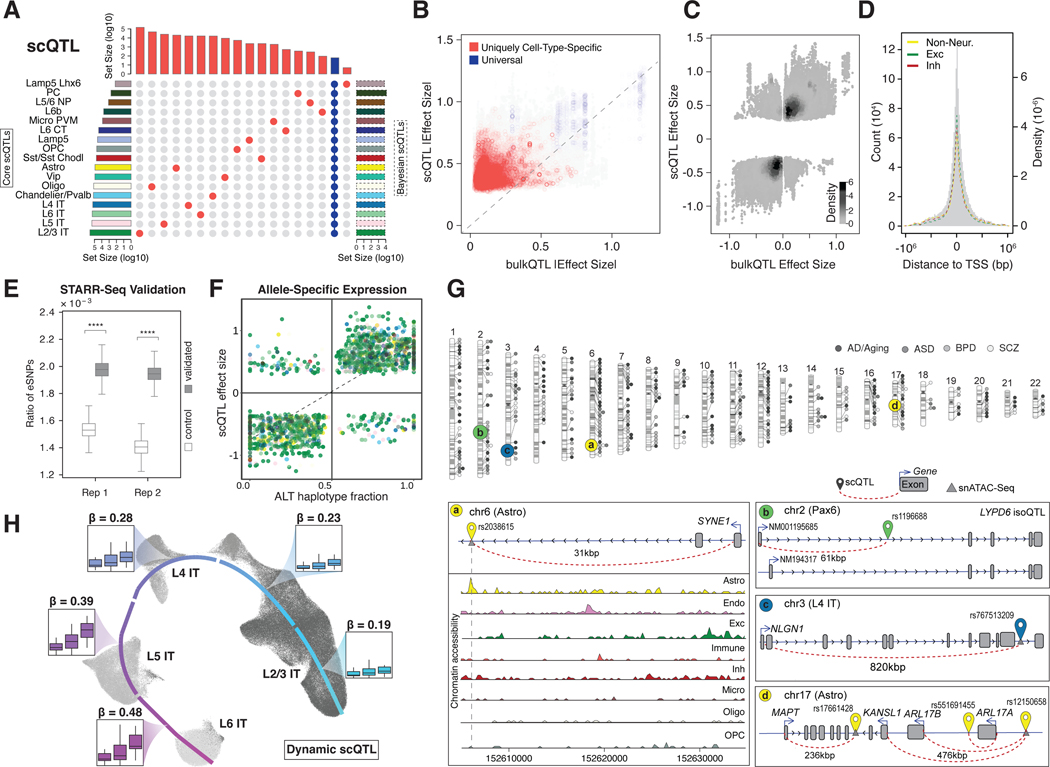
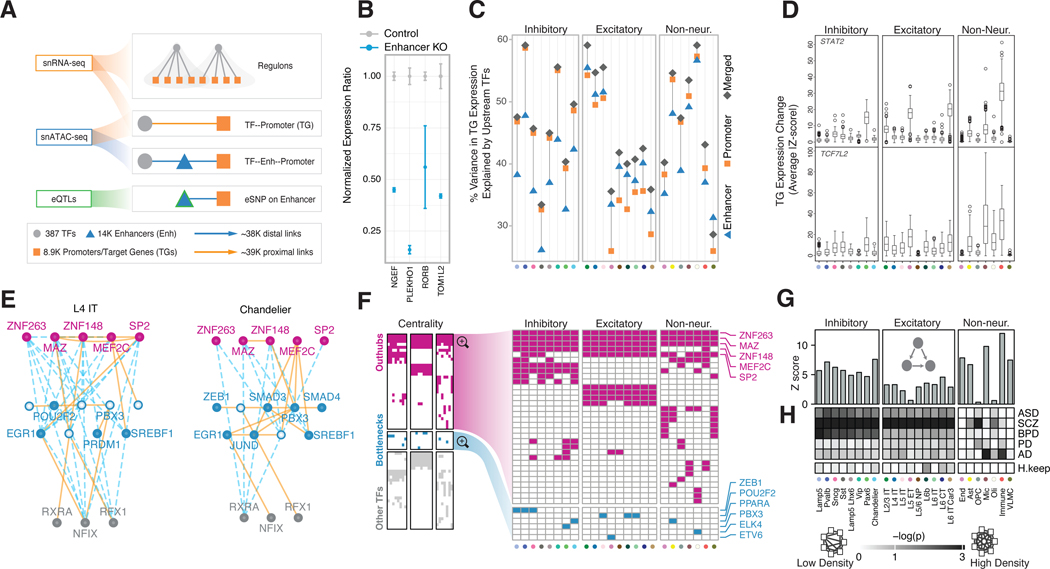
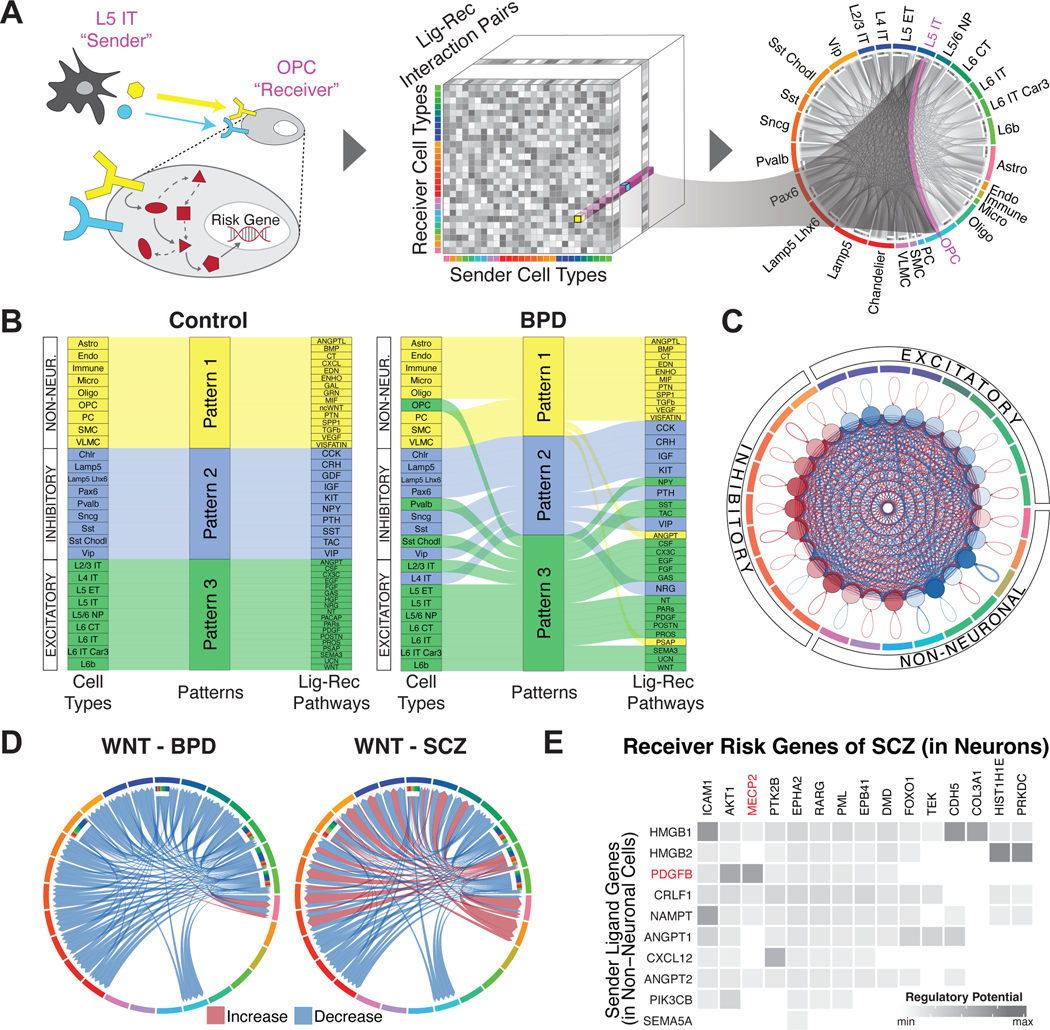
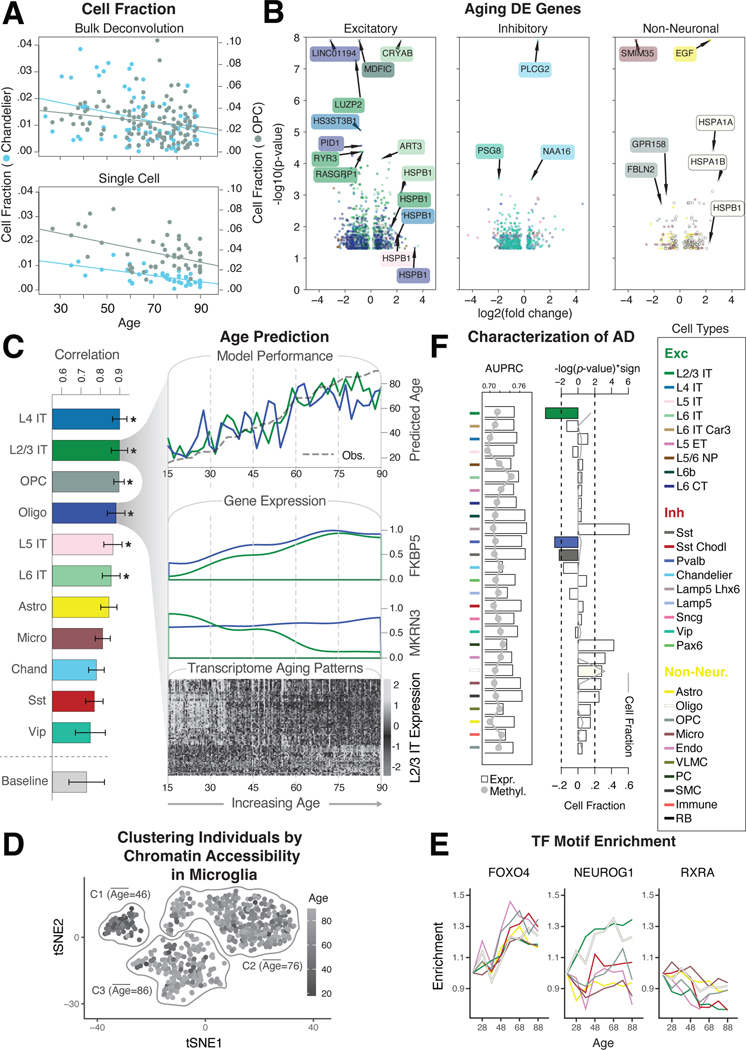
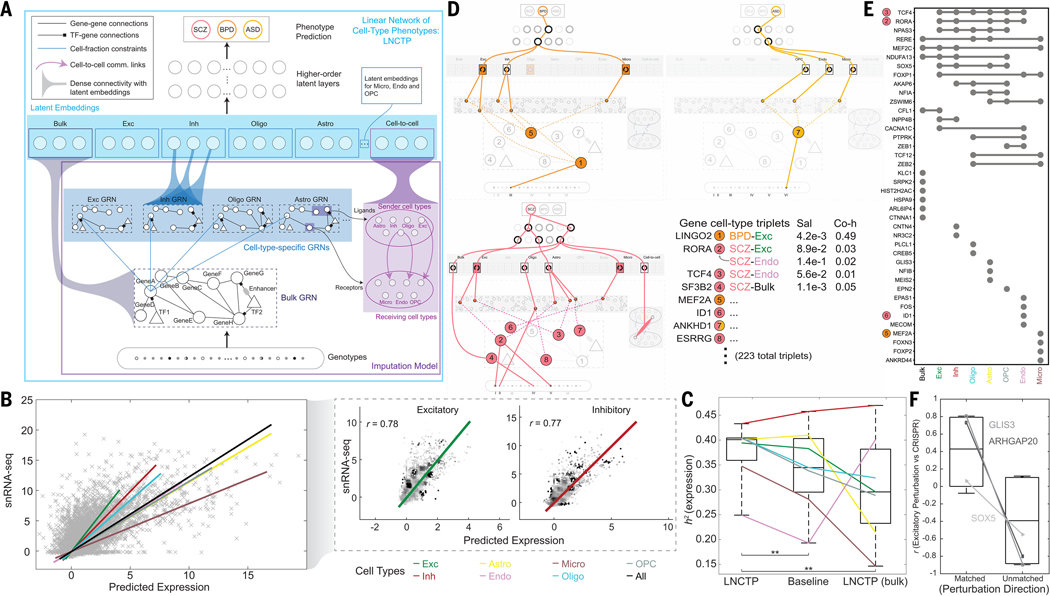
Single-cell genomics is a powerful tool for studying heterogeneous tissues such as the brain. Yet little is understood about how genetic variants influence cell-level gene expression. Addressing this, we uniformly processed single-nuclei, multiomics datasets into a resource comprising >2.8 million nuclei from the prefrontal cortex across 388 individuals. For 28 cell types, we assessed population-level variation in expression and chromatin across gene families and drug targets. We identified >550,000 cell type-specific regulatory elements and >1.4 million single-cell expression quantitative trait loci, which we used to build cell-type regulatory and cell-to-cell communication networks. These networks manifest cellular changes in aging and neuropsychiatric disorders. We further constructed an integrative model accurately imputing single-cell expression and simulating perturbations; the model prioritized ~250 disease-risk genes and drug targets with associated cell types.
Conflict of interest statement
Competing interests
Z. Weng (UMass Chan Medical School) co-founded and serves as a scientific advisor for Rgenta Inc. From April 11, 2022, N.L. Jorstad (Allen Institute for Brain Science) has been an employee of Genentech. K.P.W. (National University of Singapore) is a shareholder in Tempus AI and Provaxus Inc. The other authors declare no competing interests.
Figures
Update of
-
Single-cell genomics and regulatory networks for 388 human brains.bioRxiv [Preprint]. 2024 Mar 30:2024.03.18.585576. doi: 10.1101/2024.03.18.585576. bioRxiv. 2024. Update in: Science. 2024 May 24;384(6698):eadi5199. doi: 10.1126/science.adi5199. PMID: 38562822 Free PMC article. Updated. Preprint.
References
-
- PsychENCODE Consortium, Revealing the brain’s molecular architecture. Science 362, 1262–1263 (2018). - PubMed
MeSH terms
Substances
Grants and funding
- R01 AG050986/AG/NIA NIH HHS/United States
- R21 MH109956/MH/NIMH NIH HHS/United States
- R01 MH116529/MH/NIMH NIH HHS/United States
- U01 MH103346/MH/NIMH NIH HHS/United States
- R01 AG067025/AG/NIA NIH HHS/United States
- U01 MH116487/MH/NIMH NIH HHS/United States
- R01 MH126393/MH/NIMH NIH HHS/United States
- R01 MH110926/MH/NIMH NIH HHS/United States
- U01 MH116488/MH/NIMH NIH HHS/United States
- R01 MH110928/MH/NIMH NIH HHS/United States
- RF1 MH128970/MH/NIMH NIH HHS/United States
- U01 MH116438/MH/NIMH NIH HHS/United States
- U01 MH116442/MH/NIMH NIH HHS/United States
- R01 MH094714/MH/NIMH NIH HHS/United States
- R01 MH117406/MH/NIMH NIH HHS/United States
- R01 MH109897/MH/NIMH NIH HHS/United States
- U01 MH122591/MH/NIMH NIH HHS/United States
- R21 NS127432/NS/NINDS NIH HHS/United States
- R56 MH114901/MH/NIMH NIH HHS/United States
- R01 MH117292/MH/NIMH NIH HHS/United States
- I01 BX005160/BX/BLRD VA/United States
- T32 GM008042/GM/NIGMS NIH HHS/United States
- R21 MH103877/MH/NIMH NIH HHS/United States
- U01 MH114812/MH/NIMH NIH HHS/United States
- U01 MH103365/MH/NIMH NIH HHS/United States
- R01 MH110905/MH/NIMH NIH HHS/United States
- U01 MH122509/MH/NIMH NIH HHS/United States
- U01 MH116489/MH/NIMH NIH HHS/United States
- R01 NS128523/NS/NINDS NIH HHS/United States
- R01 MH110927/MH/NIMH NIH HHS/United States
- U01 MH122678/MH/NIMH NIH HHS/United States
- U01 MH116441/MH/NIMH NIH HHS/United States
- R01 MH122681/MH/NIMH NIH HHS/United States
- R01 MH121521/MH/NIMH NIH HHS/United States
- U01 MH122849/MH/NIMH NIH HHS/United States
- R56 MH114899/MH/NIMH NIH HHS/United States
- U01 MH103392/MH/NIMH NIH HHS/United States
- R01 MH117291/MH/NIMH NIH HHS/United States
- R21 NS128761/NS/NINDS NIH HHS/United States
- U01 MH122590/MH/NIMH NIH HHS/United States
- R01 MH117293/MH/NIMH NIH HHS/United States
- R01 MH109677/MH/NIMH NIH HHS/United States
- R56 MH114911/MH/NIMH NIH HHS/United States
- R01 MH105472/MH/NIMH NIH HHS/United States
- R01 MH129301/MH/NIMH NIH HHS/United States
- R01 MH111721/MH/NIMH NIH HHS/United States
- P50 HD105353/HD/NICHD NIH HHS/United States
- R01 MH126459/MH/NIMH NIH HHS/United States
- U01 MH103340/MH/NIMH NIH HHS/United States
- U19 AG060909/AG/NIA NIH HHS/United States
- U01 MH103339/MH/NIMH NIH HHS/United States
- I01 BX002395/BX/BLRD VA/United States
- U01 DA048279/DA/NIDA NIH HHS/United States
- R01 MH110920/MH/NIMH NIH HHS/United States
- U01 MH122681/MH/NIMH NIH HHS/United States
- R21 MH102791/MH/NIMH NIH HHS/United States
- R01 MH105898/MH/NIMH NIH HHS/United States
- R21 MH105881/MH/NIMH NIH HHS/United States
- R01 AG065582/AG/NIA NIH HHS/United States
- U01 MH122592/MH/NIMH NIH HHS/United States
- R01 MH125516/MH/NIMH NIH HHS/United States
- R01 MH125246/MH/NIMH NIH HHS/United States
- R01 MH110921/MH/NIMH NIH HHS/United States
- R01 MH123922/MH/NIMH NIH HHS/United States
- R21 MH129817/MH/NIMH NIH HHS/United States
- R01 MH109715/MH/NIMH NIH HHS/United States
- R01 HG012572/HG/NHGRI NIH HHS/United States
- RF1 MH128695/MH/NIMH NIH HHS/United States
- P30 AG066514/AG/NIA NIH HHS/United States
- R21 MH105853/MH/NIMH NIH HHS/United States
- U01 MH116492/MH/NIMH NIH HHS/United States
- P50 MH106934/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
