

A data-driven single-cell and spatial transcriptomic map of the human prefrontal cortex
- PMID: 38781370
- PMCID: PMC11398705
- DOI: 10.1126/science.adh1938
A data-driven single-cell and spatial transcriptomic map of the human prefrontal cortex
Abstract
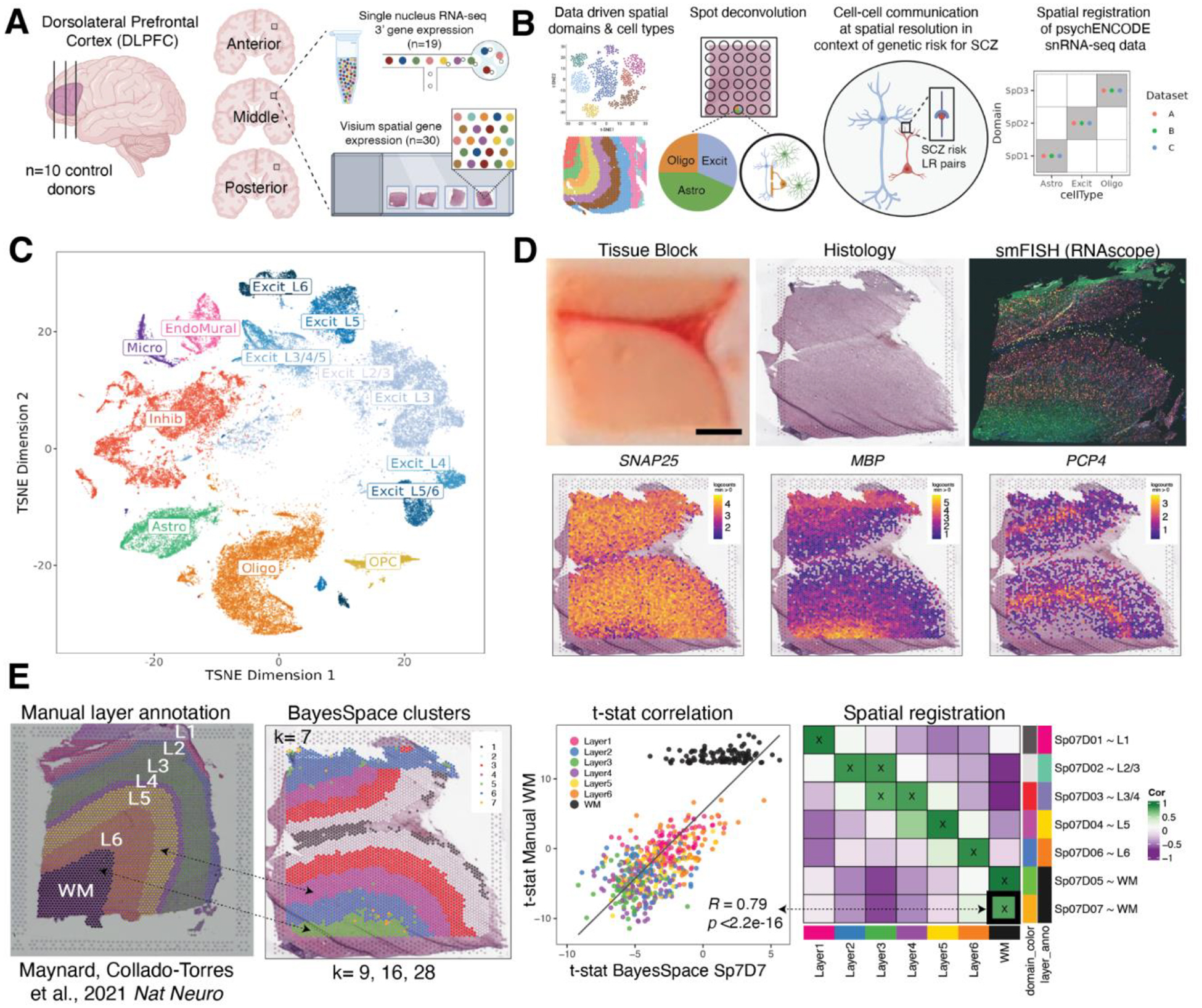
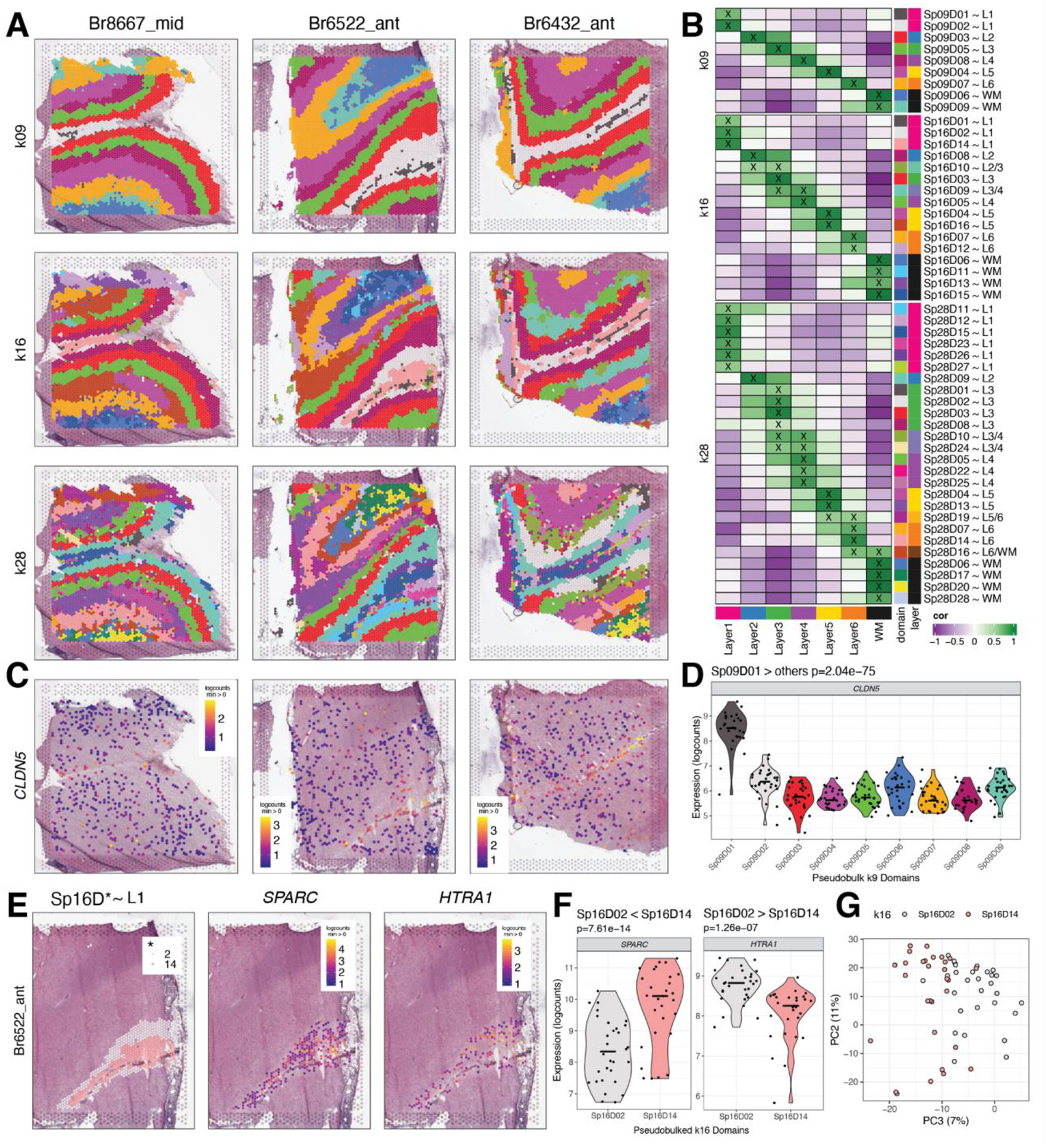
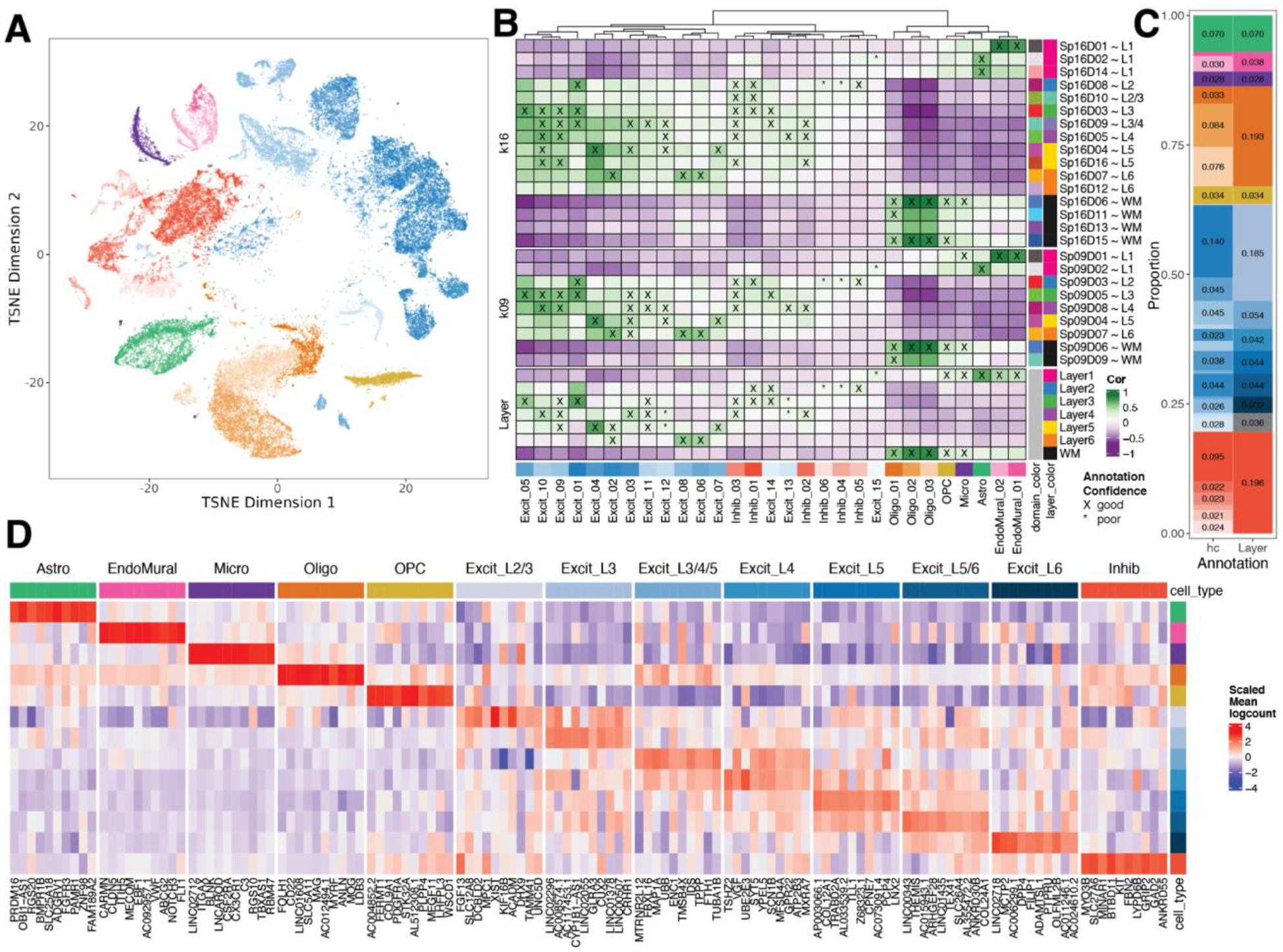
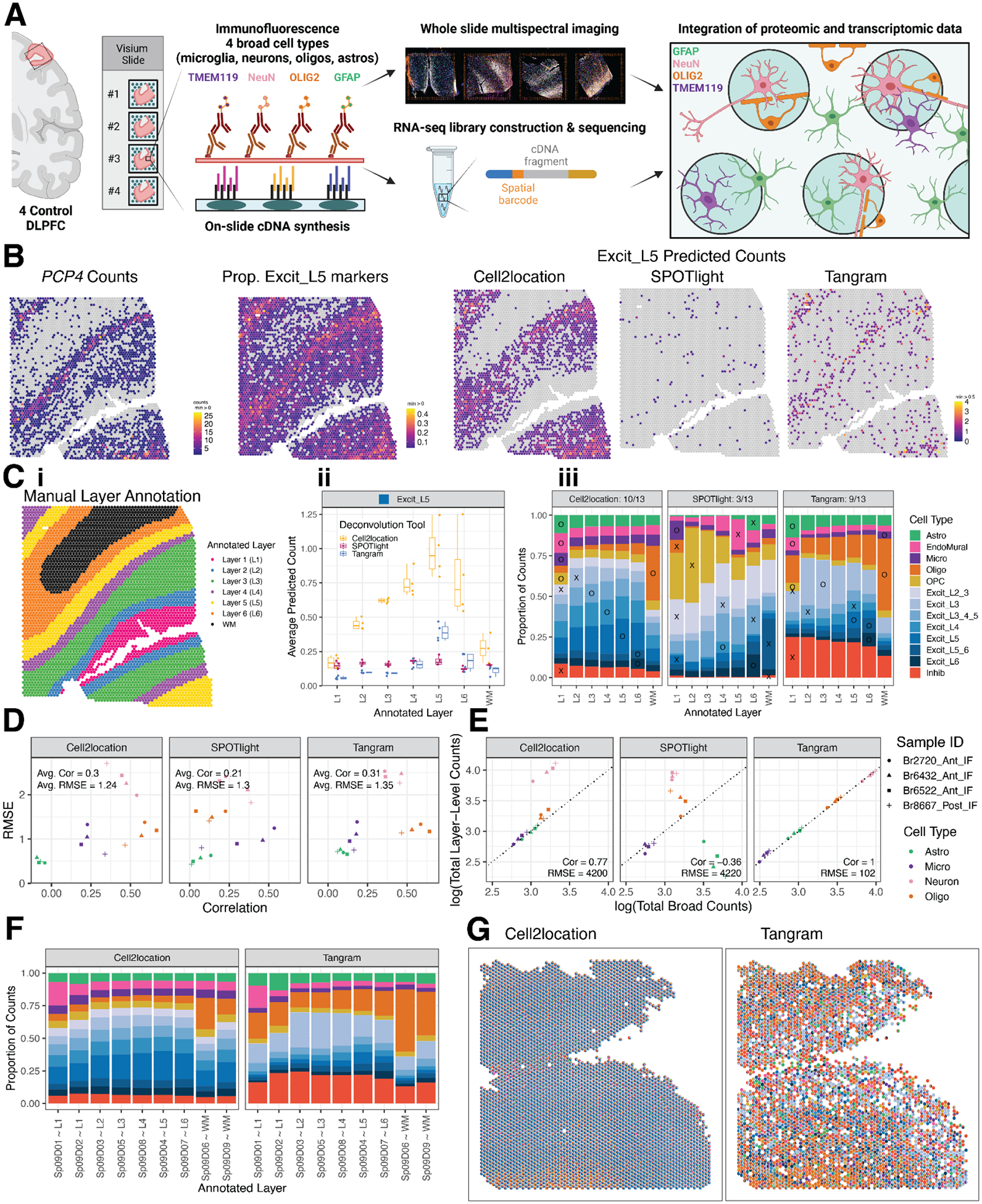
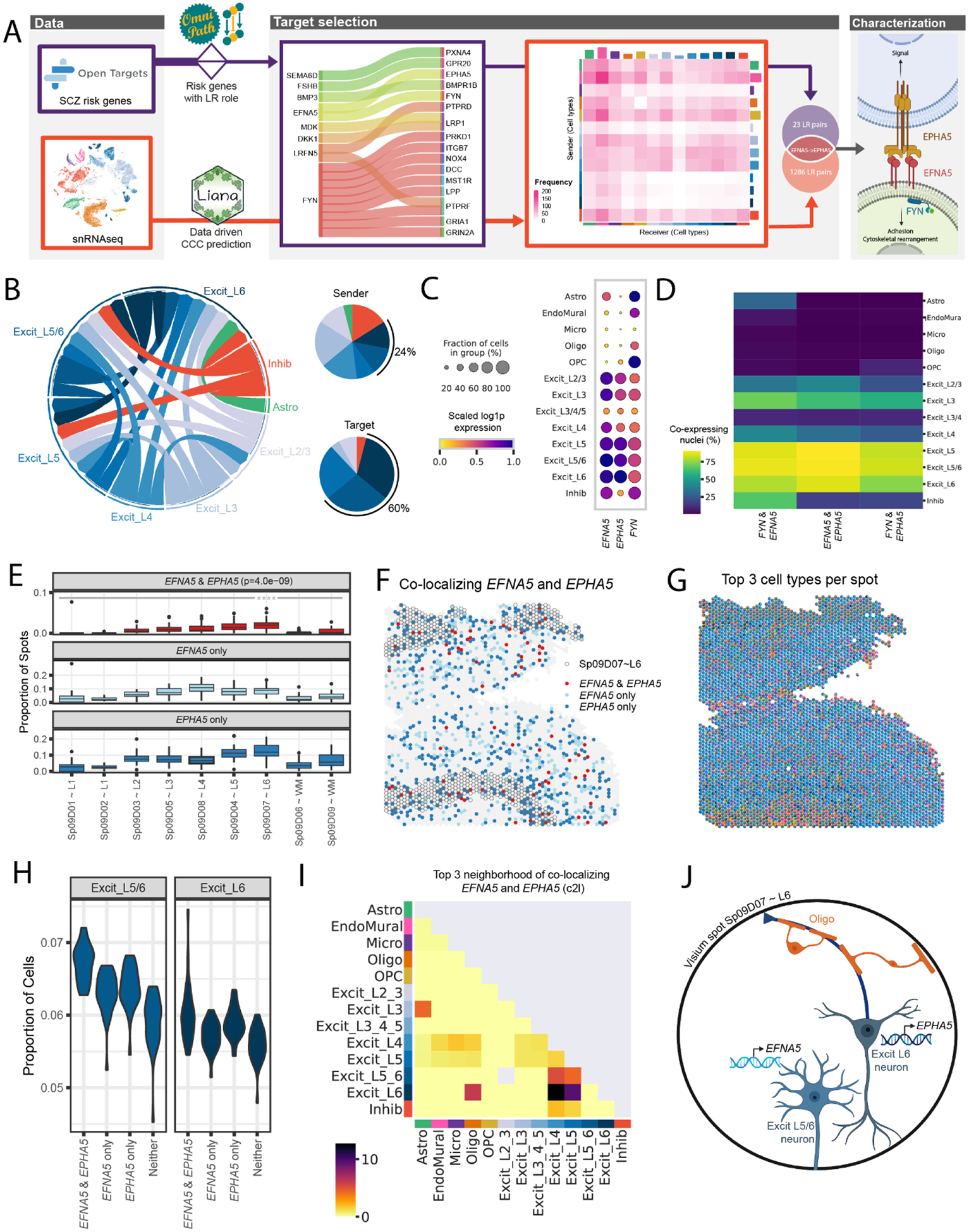
The molecular organization of the human neocortex historically has been studied in the context of its histological layers. However, emerging spatial transcriptomic technologies have enabled unbiased identification of transcriptionally defined spatial domains that move beyond classic cytoarchitecture. We used the Visium spatial gene expression platform to generate a data-driven molecular neuroanatomical atlas across the anterior-posterior axis of the human dorsolateral prefrontal cortex. Integration with paired single-nucleus RNA-sequencing data revealed distinct cell type compositions and cell-cell interactions across spatial domains. Using PsychENCODE and publicly available data, we mapped the enrichment of cell types and genes associated with neuropsychiatric disorders to discrete spatial domains.
Conflict of interest statement
Competing Interests
AB was a consultant for Third Rock Ventures and a shareholder in Alphabet, Inc. Joel E. Kleinman is a consultant on a Data Monitoring Committee for an antipsychotic drug trial for Merck & Co., Inc.
Figures

Update of
-
Integrated single cell and unsupervised spatial transcriptomic analysis defines molecular anatomy of the human dorsolateral prefrontal cortex.bioRxiv [Preprint]. 2023 Feb 15:2023.02.15.528722. doi: 10.1101/2023.02.15.528722. bioRxiv. 2023. Update in: Science. 2024 May 24;384(6698):eadh1938. doi: 10.1126/science.adh1938. PMID: 36824961 Free PMC article. Updated. Preprint.
References
-
- Madissoon E, Oliver AJ, Kleshchevnikov V, Wilbrey-Clark A, Polanski K, Richoz N, Ribeiro Orsi A, Mamanova L, Bolt L, Elmentaite R, Pett JP, Huang N, Xu C, He P, Dabrowska M, Pritchard S, Tuck L, Prigmore E, Perera S, Knights A, Meyer KB, A spatially resolved atlas of the human lung characterizes a gland-associated immune niche. Nat. Genet 55, 66–77 (2023). - PMC - PubMed
-
- Asp M, Giacomello S, Larsson L, Wu C, Fürth D, Qian X, Wärdell E, Custodio J, Reimegård J, Salmén F, Österholm C, Ståhl PL, Sundström E, Åkesson E, Bergmann O, Bienko M, Månsson-Broberg A, Nilsson M, Sylvén C, Lundeberg J, A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell. 179, 1647–1660.e19 (2019). - PubMed
-
- Hildebrandt F, Andersson A, Saarenpää S, Larsson L, Van Hul N, Kanatani S, Masek J, Ellis E, Barragan A, Mollbrink A, Andersson ER, Lundeberg J, Ankarklev J, Spatial Transcriptomics to define transcriptional patterns of zonation and structural components in the mouse liver. Nat. Commun 12, 7046 (2021). - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- R01 MH110928/MH/NIMH NIH HHS/United States
- U01 MH103365/MH/NIMH NIH HHS/United States
- U01 MH116489/MH/NIMH NIH HHS/United States
- R21 MH109956/MH/NIMH NIH HHS/United States
- R01 MH116529/MH/NIMH NIH HHS/United States
- U01 MH103346/MH/NIMH NIH HHS/United States
- U01 MH103339/MH/NIMH NIH HHS/United States
- U01 MH116487/MH/NIMH NIH HHS/United States
- R01 MH126393/MH/NIMH NIH HHS/United States
- R01 MH110926/MH/NIMH NIH HHS/United States
- U01 MH116488/MH/NIMH NIH HHS/United States
- U01 MH116438/MH/NIMH NIH HHS/United States
- U01 MH116442/MH/NIMH NIH HHS/United States
- R01 MH094714/MH/NIMH NIH HHS/United States
- P30 ES010126/ES/NIEHS NIH HHS/United States
- R01 MH117406/MH/NIMH NIH HHS/United States
- R56 MH114901/MH/NIMH NIH HHS/United States
- U01 MH122591/MH/NIMH NIH HHS/United States
- R01 MH117292/MH/NIMH NIH HHS/United States
- R21 MH103877/MH/NIMH NIH HHS/United States
- R01 MH110905/MH/NIMH NIH HHS/United States
- U01 MH122509/MH/NIMH NIH HHS/United States
- R01 MH110927/MH/NIMH NIH HHS/United States
- U01 MH122678/MH/NIMH NIH HHS/United States
- U01 MH116441/MH/NIMH NIH HHS/United States
- R01 MH121521/MH/NIMH NIH HHS/United States
- U01 MH122849/MH/NIMH NIH HHS/United States
- R56 MH114899/MH/NIMH NIH HHS/United States
- U01 MH103392/MH/NIMH NIH HHS/United States
- R01 MH117291/MH/NIMH NIH HHS/United States
- U01 MH122590/MH/NIMH NIH HHS/United States
- R01 MH117293/MH/NIMH NIH HHS/United States
- R01 MH109677/MH/NIMH NIH HHS/United States
- R56 MH114911/MH/NIMH NIH HHS/United States
- R01 MH105472/MH/NIMH NIH HHS/United States
- R01 MH129301/MH/NIMH NIH HHS/United States
- R01 MH111721/MH/NIMH NIH HHS/United States
- R01 MH126459/MH/NIMH NIH HHS/United States
- U01 MH103340/MH/NIMH NIH HHS/United States
- U01 DA048279/DA/NIDA NIH HHS/United States
- R01 MH110920/MH/NIMH NIH HHS/United States
- U01 MH122681/MH/NIMH NIH HHS/United States
- R01 MH123183/MH/NIMH NIH HHS/United States
- R21 MH102791/MH/NIMH NIH HHS/United States
- R01 MH105898/MH/NIMH NIH HHS/United States
- R35 GM139580/GM/NIGMS NIH HHS/United States
- R21 MH105881/MH/NIMH NIH HHS/United States
- U01 MH122592/MH/NIMH NIH HHS/United States
- R01 MH125516/MH/NIMH NIH HHS/United States
- R01 MH110921/MH/NIMH NIH HHS/United States
- R21 MH129817/MH/NIMH NIH HHS/United States
- R01 MH109715/MH/NIMH NIH HHS/United States
- P30 AG066514/AG/NIA NIH HHS/United States
- R21 MH105853/MH/NIMH NIH HHS/United States
- U01 MH116492/MH/NIMH NIH HHS/United States
- P50 MH106934/MH/NIMH NIH HHS/United States
