

Pathobiological signatures of dysbiotic lung injury in pediatric patients undergoing stem cell transplantation
- PMID: 38783139
- PMCID: PMC11271406
- DOI: 10.1038/s41591-024-02999-4
Pathobiological signatures of dysbiotic lung injury in pediatric patients undergoing stem cell transplantation
Abstract
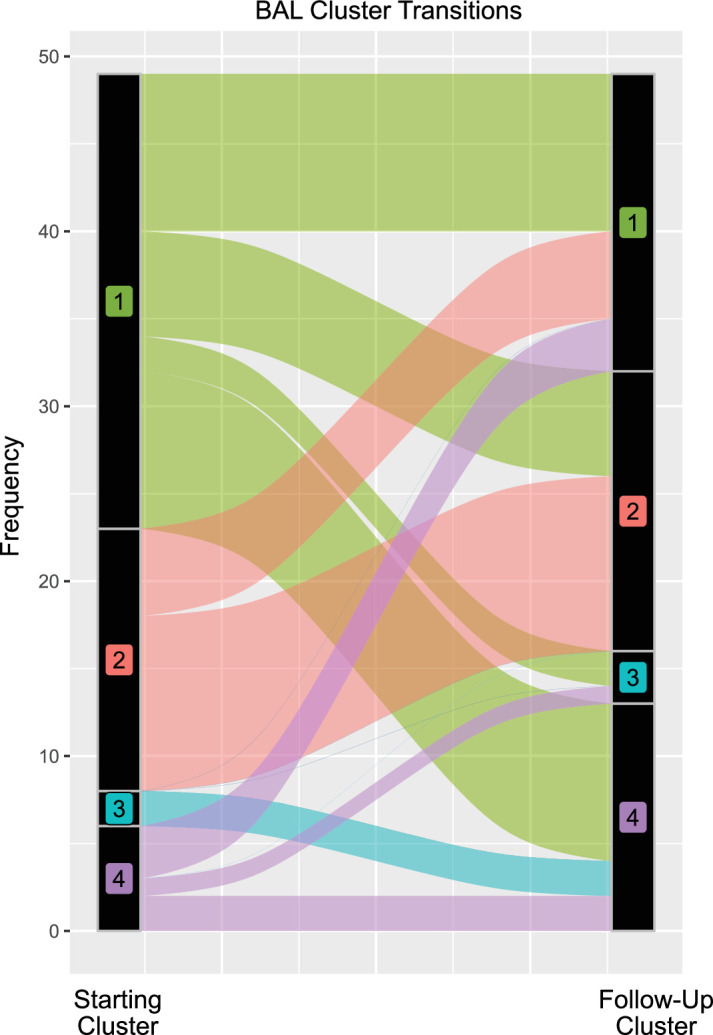
Hematopoietic cell transplantation (HCT) uses cytotoxic chemotherapy and/or radiation followed by intravenous infusion of stem cells to cure malignancies, bone marrow failure and inborn errors of immunity, hemoglobin and metabolism. Lung injury is a known complication of the process, due in part to disruption in the pulmonary microenvironment by insults such as infection, alloreactive inflammation and cellular toxicity. How microorganisms, immunity and the respiratory epithelium interact to contribute to lung injury is uncertain, limiting the development of prevention and treatment strategies. Here we used 278 bronchoalveolar lavage (BAL) fluid samples to study the lung microenvironment in 229 pediatric patients who have undergone HCT treated at 32 children's hospitals between 2014 and 2022. By leveraging paired microbiome and human gene expression data, we identified high-risk BAL compositions associated with in-hospital mortality (P = 0.007). Disadvantageous profiles included bacterial overgrowth with neutrophilic inflammation, microbiome contraction with epithelial fibroproliferation and profound commensal depletion with viral and staphylococcal enrichment, lymphocytic activation and cellular injury, and were replicated in an independent cohort from the Netherlands (P = 0.022). In addition, a broad array of previously occult pathogens was identified, as well as a strong link between antibiotic exposure, commensal bacterial depletion and enrichment of viruses and fungi. Together these lung-immune system-microorganism interactions clarify the important drivers of fatal lung injury in pediatric patients who have undergone HCT. Further investigation is needed to determine how personalized interpretation of heterogeneous pulmonary microenvironments may be used to improve pediatric HCT outcomes.
© 2024. The Author(s).
Conflict of interest statement
M.S.Z. has carried out consulting and advisory board work for Sobi. C.C.D. has carried out consulting and advisory board work for Jazz Pharmaceuticals and Alexion. J.J.A. has carried out consulting and advisory board work for AscellaHealth and Takeda. T.C.Q. has carried out consulting and advisory board work for Alexion, AstraZeneca Rare Disease and Jazz Pharmaceuticals. H.A-A. has provided research support for Adaptive. R.P. has carried out consulting and advisory board work for BlueBird Bio and provided research support to Amgen. M.A.P. has carried out consulting and advisory board work for Novartis, Pfizer, Cargo, BlueBird Bio and Vertex, and provided research support to Miltenyi Biotec and Adaptive. L.N.S. has carried out consulting and advisory board work for Sanofi. J.J.B. has carried out consulting and advisory board work for Sanofi, BlueRock, Sobi, SmartImmune, Immusoft, Advanced Clinical and Merck. J.L.D. has received salary and research support from the Chan Zuckerberg Biohub Network.
Figures













References
-
- Zinter, M. S. et al. Comprehensive prognostication in critically ill pediatric hematopoietic cell transplant patients: results from merging the Center for International Blood and Marrow Transplant Research (CIBMTR) and Virtual Pediatric Systems (VPS) registries. Biol. Blood Marrow Transplant.26, 333–342 (2020). 10.1016/j.bbmt.2019.09.027 - DOI - PMC - PubMed
MeSH terms
Grants and funding
- UG1HL069254/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- U2CCA271890/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R33 GM147800/GM/NIGMS NIH HHS/United States
- R21 GM147800/GM/NIGMS NIH HHS/United States
- K12HD000850/U.S. Department of Health & Human Services | NIH | Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD)
- K12 HD000850/HD/NICHD NIH HHS/United States
- P30CA040214/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R37 CA244775/CA/NCI NIH HHS/United States
- UG1 HL069254/HL/NHLBI NIH HHS/United States
- P30 CA042014/CA/NCI NIH HHS/United States
- P30CA008748/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- U2C CA271890/CA/NCI NIH HHS/United States
- K23 HL146936/HL/NHLBI NIH HHS/United States
- F31CA271571/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- P30 CA008748/CA/NCI NIH HHS/United States
- Foundation grant/American Thoracic Society (American Thoracic Society, Inc.)
- R37CA244775/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- K23HL146936/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R21GM147800/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- F31 CA271571/CA/NCI NIH HHS/United States
