

Hybridization alters red deer gut microbiome and metabolites
- PMID: 38784815
- PMCID: PMC11112572
- DOI: 10.3389/fmicb.2024.1387957
Hybridization alters red deer gut microbiome and metabolites
Abstract
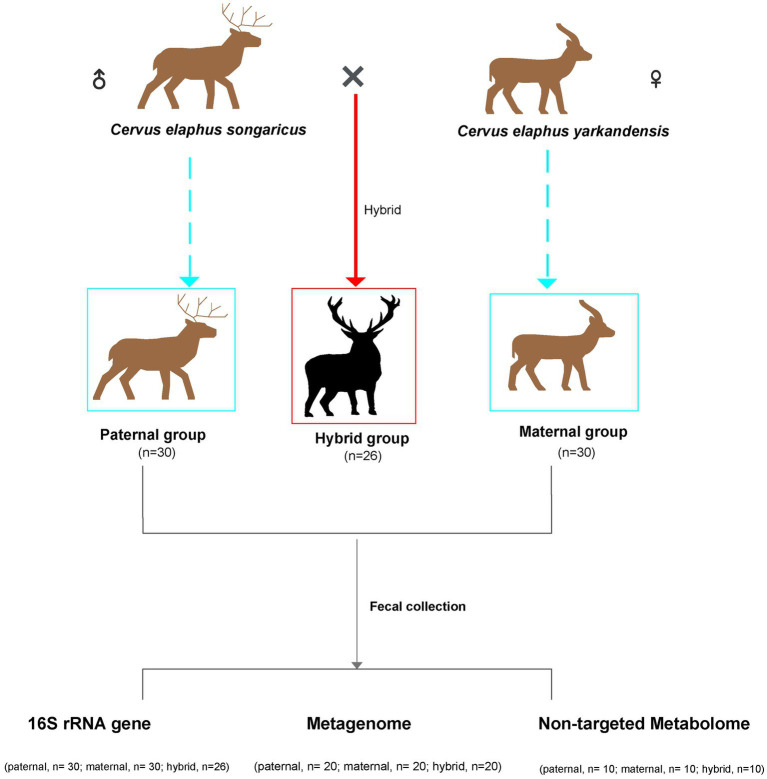
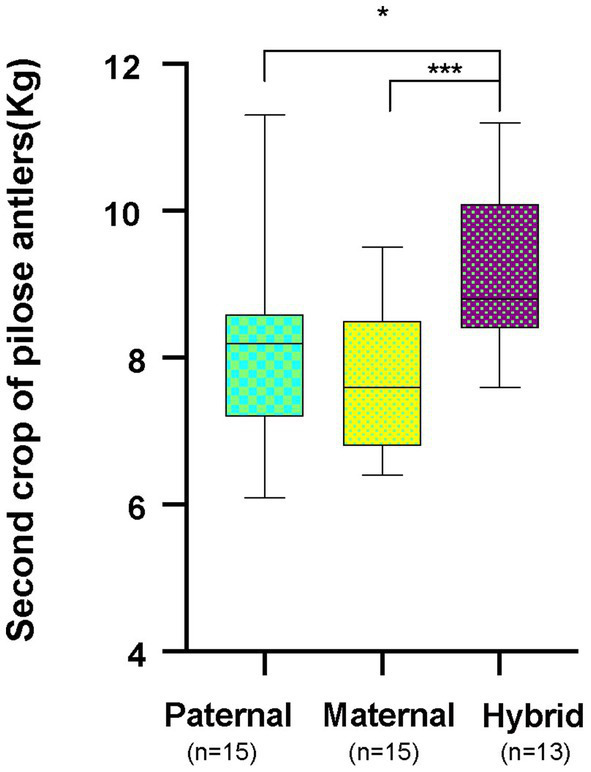
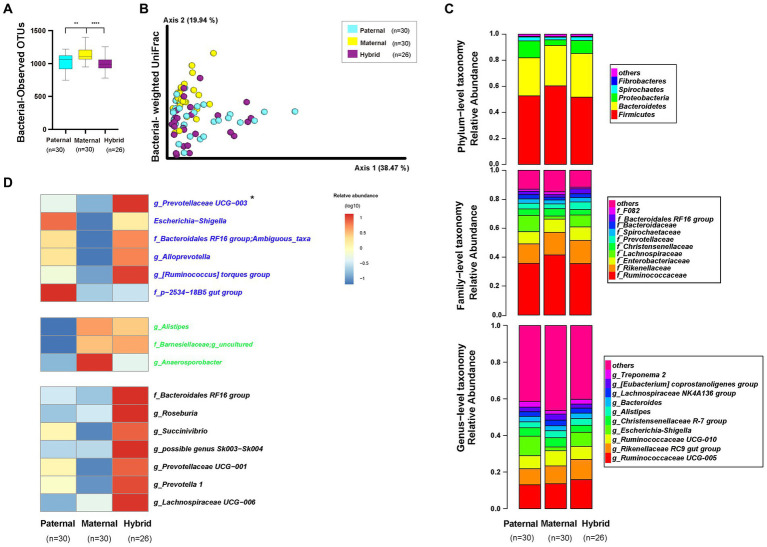
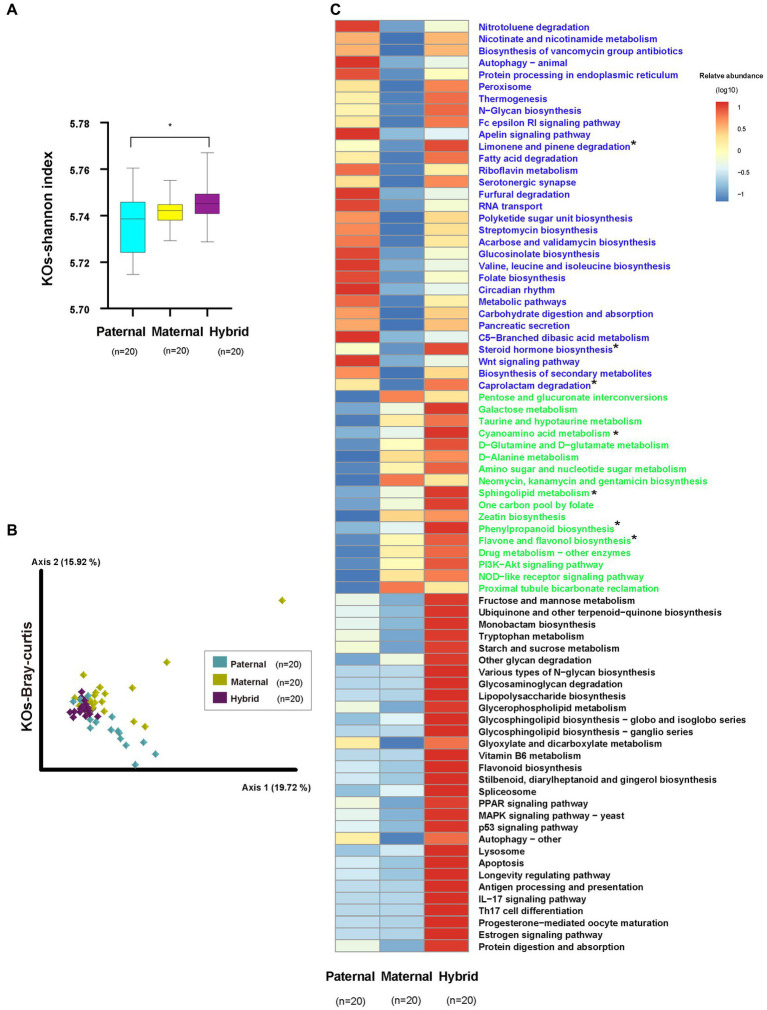
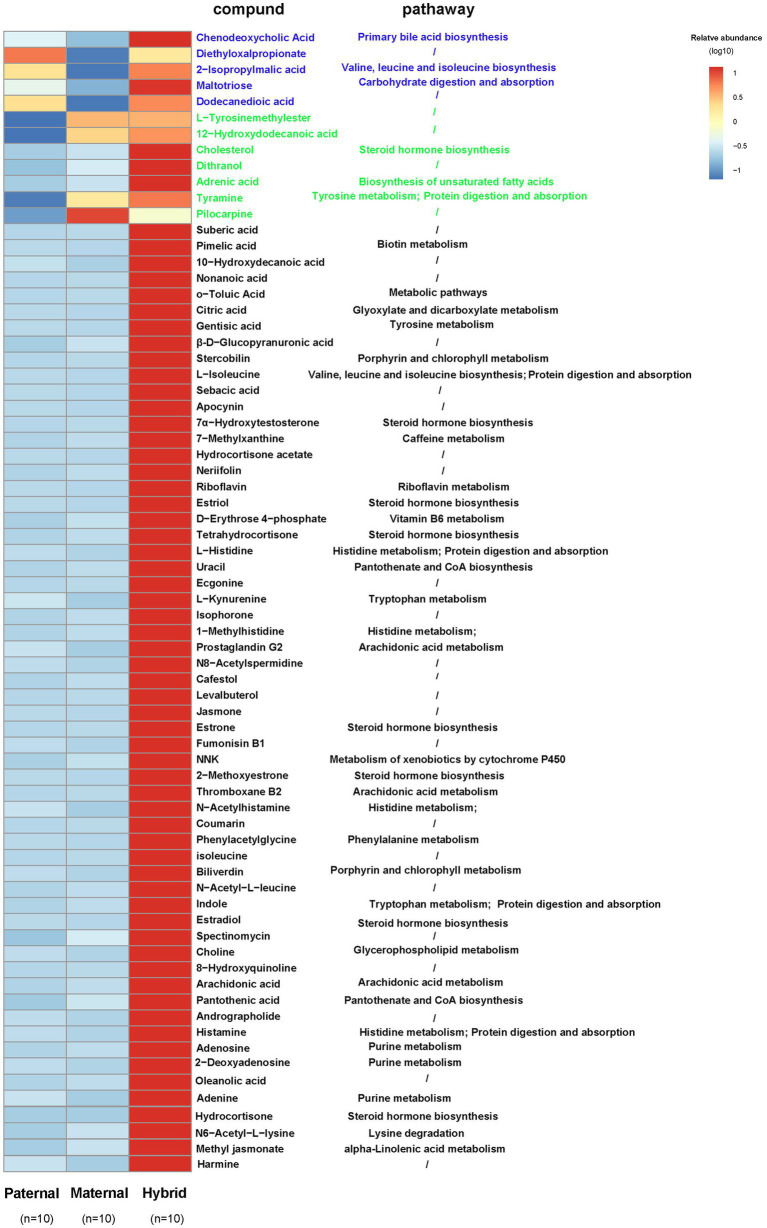
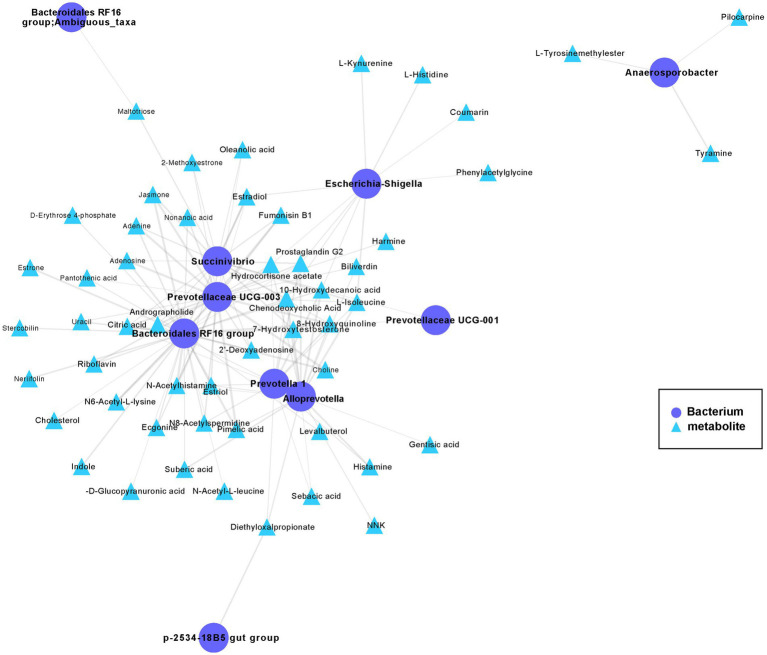
The host genes play a crucial role in shaping the composition and structure of the gut microbiome. Red deer is listed as an endangered species by the International Union for the Conservation of Nature, and its pilose antlers have good medicinal value. Hybridization can lead to heterosis, resulting in increased pilose antler production and growth performance in hybrid deer. However, the role of the gut microbiome in hybrid deer remains largely unknown. In this study, alpha and beta diversity analysis showed that hybridization altered the composition and structure of the gut microbiome of the offspring, with the composition and structure of the hybrid offspring being more similar to those of the paternal parents. Interestingly, the LefSe differential analysis showed that there were some significantly enriched gut microbiome in the paternal parents (such as g_Prevotellaceae UCG-003, f_Bacteroidales RF16 group; Ambiguous_taxa, etc.) and the maternal parents (including g_Alistipes, g_Anaerosporobacter, etc.), which remained significantly enriched in the hybrid offspring. Additionally, the hybrid offspring exhibited a significant advantage over the parental strains, particularly in taxa that can produce short-chain fatty acids, such as g_Prevotellaceae UCG-003, g_Roseburia, g_Succinivibrio, and g_Lachnospiraceae UCG-006. Similar to bacterial transmission, metagenomic analysis showed that some signaling pathways related to pilose antler growth ("Wnt signaling pathway," "PI3K Akt signaling pathway," "MAPK signaling pathway") were also enriched in hybrid red deer after hybridization. Furthermore, metabolomic analysis revealed that compared with the paternal and maternal parents, the hybrid offspring exhibited significant enrichment in metabolites related to "Steroid hormone biosynthesis," "Tryptophan metabolism," "Valine, leucine and isoleucine metabolism," and "Vitamin B metabolism." Notably, the metagenomic analysis also showed that these metabolic pathways were significantly enriched in hybrid deer. Finally, a correlation analysis between the gut microbiome and metabolites revealed a significant positive correlation between the enriched taxa in hybrid deer, including the Bacteroidales RF16 group, Prevotellaceae, and Succinivibrio, and metabolites, such as 7α-hydroxytestosterone, L-kynurenine, indole, L-isoleucine, and riboflavin. The study contributes valuable data toward understanding the role of the gut microbiome from red deer in hybridization and provides reference data for further screening potential probiotics and performing microbial-assisted breeding that promotes the growth of red deer pilose antlers and bodies, development, and immunity.
Keywords: gut microbiome; hybridization; multi-omics technologies; pilose antler; red deer.
Copyright © 2024 Wei, Zeng, Li, Guo, Mu, Gan and Li.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Anderson S., Côté S., Richard J., Shafer A. (2019). Genomic architecture of artificially and sexually-selected traits in a wild cervid. Bio Rxiv 841528, 1–29. doi: 10.1101/841528 - DOI
-
- Auffret M. D., Stewart R. D., Dewhurst R. J., Duthie C.-A., Watson M., Roehe R. (2020). Identification of microbial genetic capacities and potential mechanisms within the rumen microbiome explaining differences in beef cattle feed efficiency. Front. Microbiol. 11:1229. doi: 10.3389/fmicb.2020.01229, PMID: - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources