

Membrane Association of the Short Transglutaminase Type 2 Splice Variant (TG2-S) Modulates Cisplatin Resistance in a Human Hepatocellular Carcinoma (HepG2) Cell Line
- PMID: 38785527
- PMCID: PMC11119602
- DOI: 10.3390/cimb46050259
Membrane Association of the Short Transglutaminase Type 2 Splice Variant (TG2-S) Modulates Cisplatin Resistance in a Human Hepatocellular Carcinoma (HepG2) Cell Line
Abstract
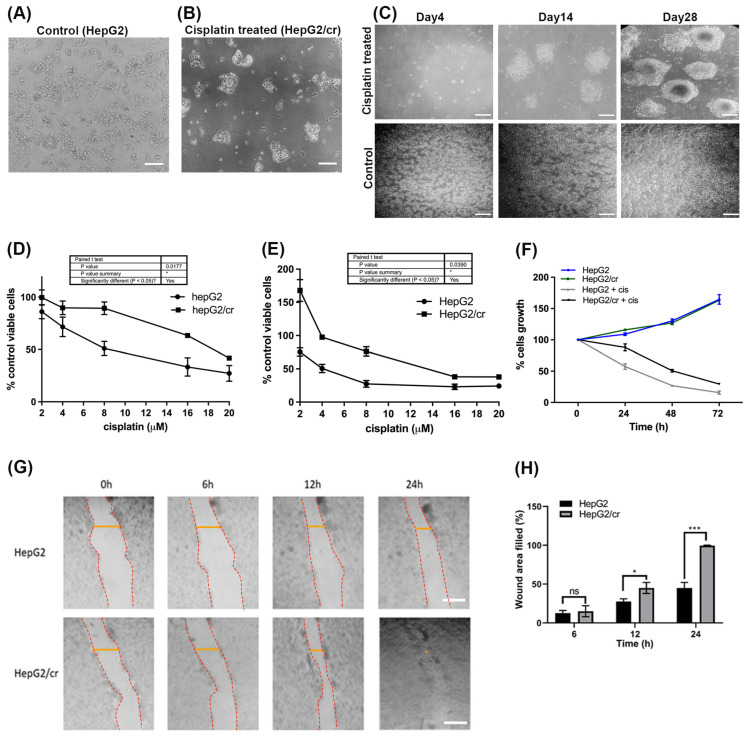
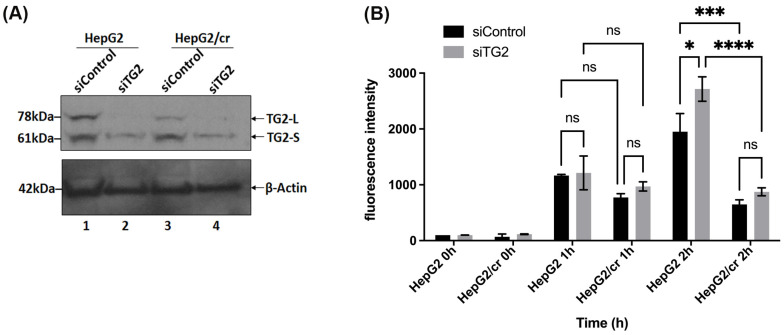
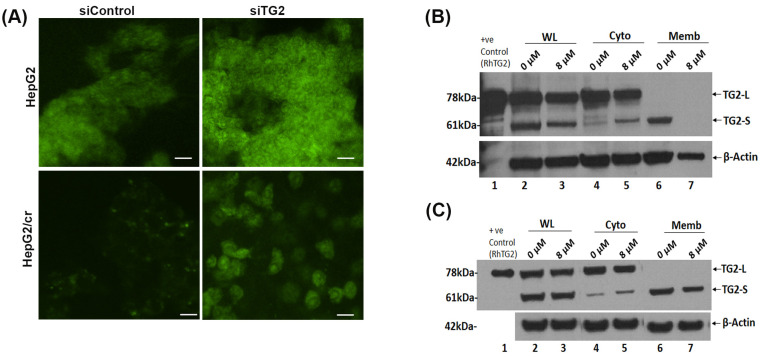
Hepatocellular carcinoma (HCC) is a heterogeneous malignancy with complex carcinogenesis. Although there has been significant progress in the treatment of HCC over the past decades, drug resistance to chemotherapy remains a major obstacle in its successful management. In this study, we were able to reduce chemoresistance in cisplatin-resistant HepG2 cells by either silencing the expression of transglutaminase type 2 (TG2) using siRNA or by the pre-treatment of cells with the TG2 enzyme inhibitor cystamine. Further analysis revealed that, whereas the full-length TG2 isoform (TG2-L) was almost completely cytoplasmic in its distribution, the majority of the short TG2 isoform (TG2-S) was membrane-associated in both parental and chemoresistant HepG2 cells. Following the induction of cisplatin toxicity in non-chemoresistant parental cells, TG2-S, together with cisplatin, quickly relocated to the cytosolic fraction. Conversely, no cytosolic relocalisation of TG2-S or nuclear accumulation cisplatin was observed, following the identical treatment of chemoresistant cells, where TG2-S remained predominantly membrane-associated. This suggests that the deficient subcellular relocalisation of TG2-S from membranous structures into the cytoplasm may limit the apoptic response to cisplatin toxicity in chemoresistant cells. Structural analysis of TG2 revealed the presence of binding motifs for interaction of TG2-S with the membrane scaffold protein LC3/LC3 homologue that could contribute to a novel mechanism of chemotherapeutic resistance in HepG2 cells.
Keywords: HepG2; apoptosis; autophagy; chemoresistance; cisplatin; hepatocellular carcinoma (HCC); sub-cellular localisation; transglutaminase 2 (TG2).
Conflict of interest statement
The authors declare no conflicts of interest.
Figures





Similar articles
-
Inhibition of Transglutaminase 2 activity increases cisplatin cytotoxicity in a model of human hepatocarcinoma chemotherapy.Eur J Pharmacol. 2017 Nov 15;815:332-342. doi: 10.1016/j.ejphar.2017.09.035. Epub 2017 Sep 22. Eur J Pharmacol. 2017. PMID: 28943101
-
Morin Hydrate Reverses Cisplatin Resistance by Impairing PARP1/HMGB1-Dependent Autophagy in Hepatocellular Carcinoma.Cancers (Basel). 2019 Jul 15;11(7):986. doi: 10.3390/cancers11070986. Cancers (Basel). 2019. PMID: 31311167 Free PMC article.
-
Tissue transglutaminase inhibits autophagy in pancreatic cancer cells.Mol Cancer Res. 2007 Mar;5(3):241-9. doi: 10.1158/1541-7786.MCR-06-0229. Mol Cancer Res. 2007. PMID: 17374730
-
Tissue transglutaminase-mediated chemoresistance in cancer cells.Drug Resist Updat. 2007 Aug-Oct;10(4-5):144-51. doi: 10.1016/j.drup.2007.06.002. Epub 2007 Jul 27. Drug Resist Updat. 2007. PMID: 17662645 Review.
-
Tissue transglutaminase promotes or suppresses tumors depending on cell context.Anticancer Res. 2009 Jun;29(6):1909-19. Anticancer Res. 2009. PMID: 19528447 Review.
Cited by
-
Retinoic Acid-Induced Transglutaminase 2 Expression Reduces Sensitivity to Cisplatin in the Hormone-Positive MCF-7 Breast Cancer Cell Model.Int J Mol Sci. 2025 Aug 21;26(16):8101. doi: 10.3390/ijms26168101. Int J Mol Sci. 2025. PMID: 40869421 Free PMC article.
References
-
- Global Burden of Disease Cancer Collaboration. Fitzmaurice C., Allen C., Barber R.M., Barregard L., Bhutta Z.A., Brenner H., Dicker D.J., Chimed-Orchir O., Dandona R., et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017;3:524–548. - PMC - PubMed
-
- Galle P.R., Forner A., Llovet J.M., Mazzaferro V., Piscaglia F., Raoul J.-L., Schirmacher P., Vilgrain V. EASL clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018;69:182–236. - PubMed
-
- Meng X.C., Chen B.H., Huang J.J., Huang W.S., Cai M.Y., Zhou J.W., Guo Y.J., Zhu K.S. Early prediction of survival in hepatocellular carcinoma patients treated with transarterial chemoembolization plus sorafenib. World J. Gastroenterol. 2018;24:484–493. doi: 10.3748/wjg.v24.i4.484. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
