

Mycoplasma glycine cleavage system key subunit GcvH is an apoptosis inhibitor targeting host endoplasmic reticulum
- PMID: 38787906
- PMCID: PMC11156438
- DOI: 10.1371/journal.ppat.1012266
Mycoplasma glycine cleavage system key subunit GcvH is an apoptosis inhibitor targeting host endoplasmic reticulum
Abstract
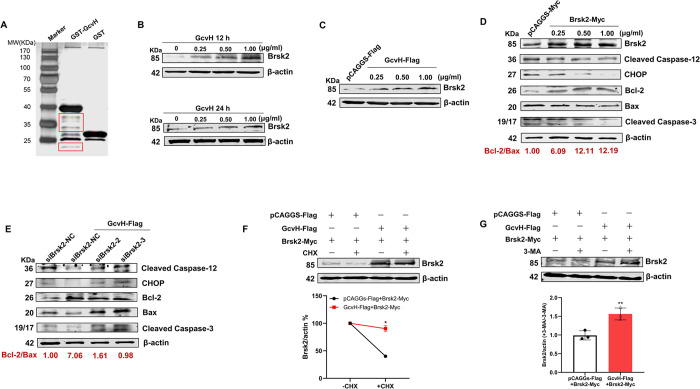
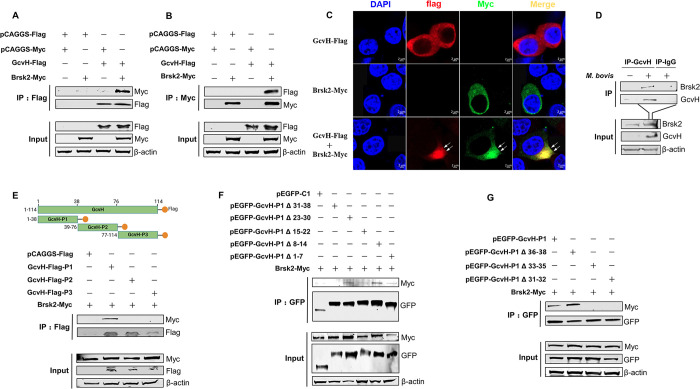
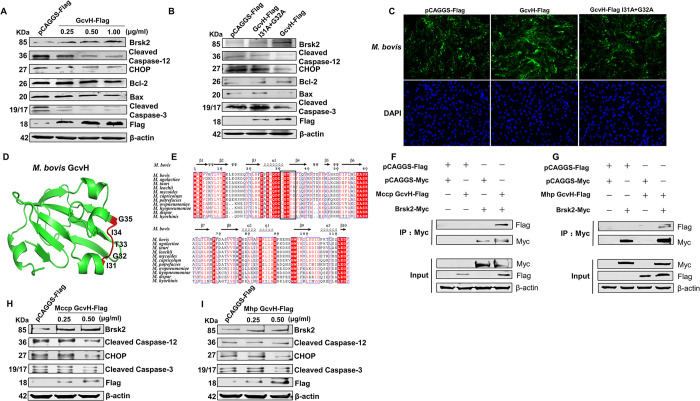
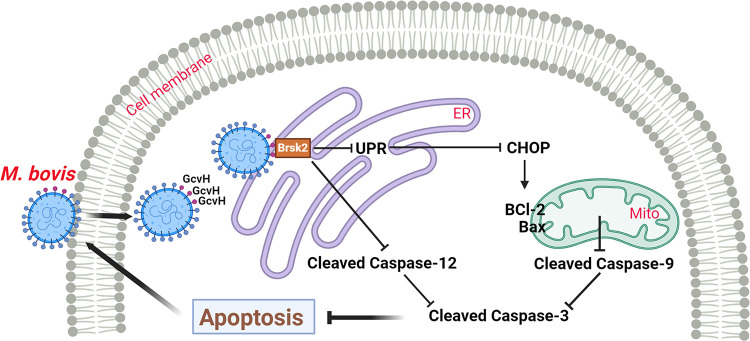
Mycoplasmas are minimal but notorious bacteria that infect humans and animals. These genome-reduced organisms have evolved strategies to overcome host apoptotic defense and establish persistent infection. Here, using Mycoplasma bovis as a model, we demonstrate that mycoplasma glycine cleavage system (GCS) H protein (GcvH) targets the endoplasmic reticulum (ER) to hijack host apoptosis facilitating bacterial infection. Mechanically, GcvH interacts with the ER-resident kinase Brsk2 and stabilizes it by blocking its autophagic degradation. Brsk2 subsequently disturbs unfolded protein response (UPR) signaling, thereby inhibiting the key apoptotic molecule CHOP expression and ER-mediated intrinsic apoptotic pathway. CHOP mediates a cross-talk between ER- and mitochondria-mediated intrinsic apoptosis. The GcvH N-terminal amino acid 31-35 region is necessary for GcvH interaction with Brsk2, as well as for GcvH to exert anti-apoptotic and potentially pro-infective functions. Notably, targeting Brsk2 to dampen apoptosis may be a conserved strategy for GCS-containing mycoplasmas. Our study reveals a novel role for the conserved metabolic route protein GcvH in Mycoplasma species. It also sheds light on how genome-reduced bacteria exploit a limited number of genomic proteins to resist host cell apoptosis thereby facilitating pathogenesis.
Copyright: © 2024 Pan et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Mechanism of the induction of endoplasmic reticulum stress by the anti-cancer agent, di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT): Activation of PERK/eIF2α, IRE1α, ATF6 and calmodulin kinase.Biochem Pharmacol. 2016 Jun 1;109:27-47. doi: 10.1016/j.bcp.2016.04.001. Epub 2016 Apr 6. Biochem Pharmacol. 2016. PMID: 27059255
-
BRSK2 is regulated by ER stress in protein level and involved in ER stress-induced apoptosis.Biochem Biophys Res Commun. 2012 Jul 13;423(4):813-8. doi: 10.1016/j.bbrc.2012.06.046. Epub 2012 Jun 16. Biochem Biophys Res Commun. 2012. PMID: 22713462
-
Mycoplasmas bovis P48 induces apoptosis in EBL cells via an endoplasmic reticulum stress-dependent signaling pathway.Vet Microbiol. 2021 Apr;255:109013. doi: 10.1016/j.vetmic.2021.109013. Epub 2021 Feb 20. Vet Microbiol. 2021. PMID: 33676093
-
Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes.Mol Metab. 2019 Sep;27S(Suppl):S60-S68. doi: 10.1016/j.molmet.2019.06.012. Mol Metab. 2019. PMID: 31500832 Free PMC article. Review.
-
The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress.Curr Mol Med. 2016;16(6):533-44. doi: 10.2174/1566524016666160523143937. Curr Mol Med. 2016. PMID: 27211800 Free PMC article. Review.
References
-
- Maes D, Sibila M, Kuhnert P, Segalés J, Haesebrouck F, Pieters M. Update on Mycoplasma hyopneumoniae infections in pigs: knowledge gaps for improved disease control. Transboundary and emerging diseases. 2018;65:110–24. - PubMed
-
- Calcutt M, Lysnyansky I, Sachse K, Fox L, Nicholas R, Ayling R. Gap analysis of Mycoplasma bovis disease, diagnosis and control: An aid to identify future development requirements. Transboundary and emerging diseases. 2018;65:91–109. - PubMed
-
- Dawood A, Algharib SA, Zhao G, Zhu T, Qi M, Delai K, et al.. Mycoplasmas as host pantropic and specific pathogens: clinical implications, gene transfer, virulence factors, and future perspectives. Frontiers in cellular and infection microbiology. 2022;12:855731. doi: 10.3389/fcimb.2022.855731 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
