

Serine Palmitoyltransferase (SPT)-related Neurodegenerative and Neurodevelopmental Disorders
- PMID: 38788085
- PMCID: PMC11307022
- DOI: 10.3233/JND-240014
Serine Palmitoyltransferase (SPT)-related Neurodegenerative and Neurodevelopmental Disorders
Abstract
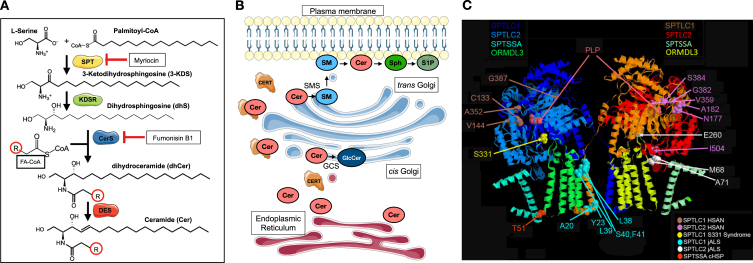
Motor neuron diseases and peripheral neuropathies are heterogeneous groups of neurodegenerative disorders that manifest with distinct symptoms due to progressive dysfunction or loss of specific neuronal subpopulations during different stages of development. A few monogenic, neurodegenerative diseases associated with primary metabolic disruptions of sphingolipid biosynthesis have been recently discovered. Sphingolipids are a subclass of lipids that form critical building blocks of all cellular and subcellular organelle membranes including the membrane components of the nervous system cells. They are especially abundant within the lipid portion of myelin. In this review, we will focus on our current understanding of disease phenotypes in three monogenic, neuromuscular diseases associated with pathogenic variants in components of serine palmitoyltransferase, the first step in sphingolipid biosynthesis. These include hereditary sensory and autonomic neuropathy type 1 (HSAN1), a sensory predominant peripheral neuropathy, and two neurodegenerative disorders: juvenile amyotrophic lateral sclerosis affecting the upper and lower motor neurons with sparing of sensory neurons, and a complicated form of hereditary spastic paraplegia with selective involvement of the upper motor neurons and more broad CNS neurodegeneration. We will also review our current understanding of disease pathomechanisms, therapeutic approaches, and the unanswered questions to explore in future studies.
Keywords: Serine palmitoyltransferase; amyotrophic lateral sclerosis; hereditary sensory and autonomic neuropathy; hereditary spastic paraplegia; sphingolipid biosynthesis.
Conflict of interest statement
PM is a consultant for Leal Therapeutics LLC (Cash only, no stock options). PM is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review. FE has received consulting fees from UpToDate, Prime Medical Education, bluebird bio, Leal Therapeutics, Sanofi, Takeda and SwanBio Therapeutics. All other authors declare no competing interests.
Figures
Similar articles
-
Recurrent de novo SPTLC2 variant causes childhood-onset amyotrophic lateral sclerosis (ALS) by excess sphingolipid synthesis.J Neurol Neurosurg Psychiatry. 2024 Jan 11;95(2):103-113. doi: 10.1136/jnnp-2023-332132. J Neurol Neurosurg Psychiatry. 2024. PMID: 38041679 Free PMC article.
-
SPTLC1 variants associated with ALS produce distinct sphingolipid signatures through impaired interaction with ORMDL proteins.J Clin Invest. 2022 Sep 15;132(18):e161908. doi: 10.1172/JCI161908. J Clin Invest. 2022. PMID: 35900868 Free PMC article.
-
SPTSSA variants alter sphingolipid synthesis and cause a complex hereditary spastic paraplegia.Brain. 2023 Apr 19;146(4):1420-1435. doi: 10.1093/brain/awac460. Brain. 2023. PMID: 36718090 Free PMC article.
-
Recent advances in hereditary sensory and autonomic neuropathies.Curr Opin Neurol. 2006 Oct;19(5):474-80. doi: 10.1097/01.wco.0000245370.82317.f6. Curr Opin Neurol. 2006. PMID: 16969157 Review.
-
Roles of l-serine and sphingolipid synthesis in brain development and neuronal survival.Prog Lipid Res. 2008 May;47(3):188-203. doi: 10.1016/j.plipres.2008.01.003. Epub 2008 Feb 15. Prog Lipid Res. 2008. PMID: 18319065 Review.
References
-
- Arenz C. Recent advances and novel treatments for sphingolipidoses. Future Med Chem. 2017;9(14):1687–700. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
