

In vitro generation of genetic diversity for directed evolution by error-prone artificial DNA synthesis
- PMID: 38789612
- PMCID: PMC11126579
- DOI: 10.1038/s42003-024-06340-0
In vitro generation of genetic diversity for directed evolution by error-prone artificial DNA synthesis
Abstract
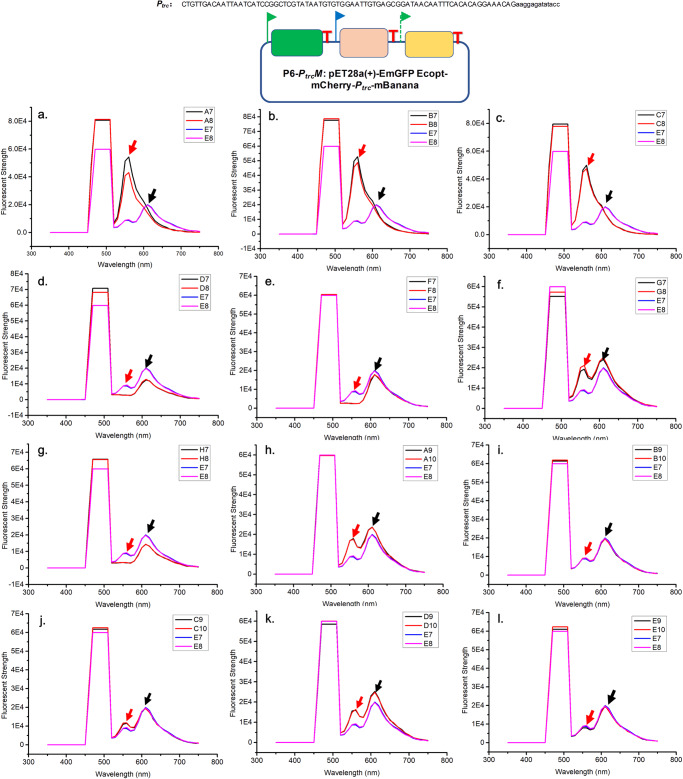
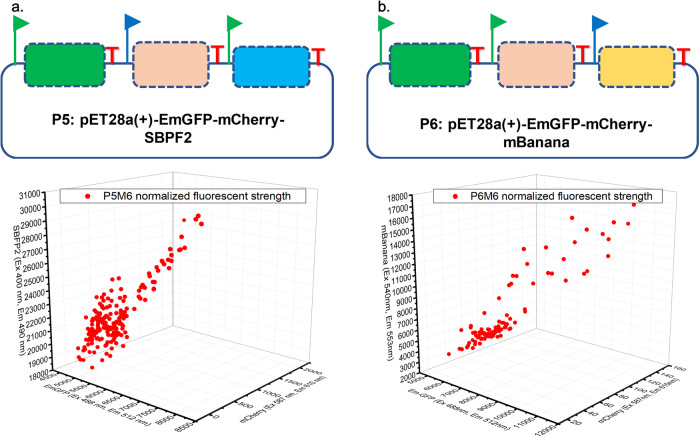
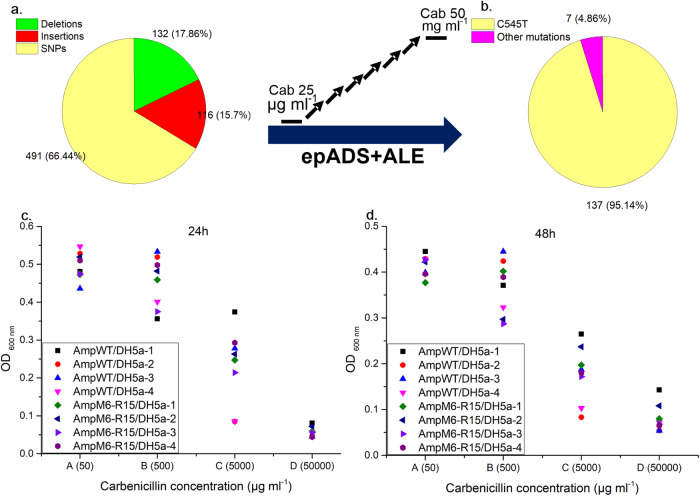
Generating genetic diversity lies at the heart of directed evolution which has been widely used to engineer genetic parts and gene circuits in synthetic biology. With the ever-expanding application of directed evolution, different approaches of generating genetic diversity are required to enrich the traditional toolbox. Here we show in vitro generation of genetic diversity for directed evolution by error-prone artificial DNA synthesis (epADS). This approach comprises a three-step process which incorporates base errors randomly generated during chemical synthesis of oligonucleotides under specific conditions into the target DNA. Through this method, 200 ~ 4000 folds of diversification in fluorescent strength have been achieved in genes encoding fluorescent proteins. EpADS has also been successfully used to diversify regulatory genetic parts, synthetic gene circuits and even increase microbial tolerance to carbenicillin in a short time period. EpADS would be an alternative tool for directed evolution which may have useful applications in synthetic biology.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
Combinatorial Evolution of DNA with RECODE.Methods Mol Biol. 2018;1772:205-212. doi: 10.1007/978-1-4939-7795-6_11. Methods Mol Biol. 2018. PMID: 29754230
-
Directed evolution of promoters and tandem gene arrays for customizing RNA synthesis rates and regulation.Methods Enzymol. 2011;497:135-55. doi: 10.1016/B978-0-12-385075-1.00006-8. Methods Enzymol. 2011. PMID: 21601085
-
Fast and Flexible Synthesis of Combinatorial Libraries for Directed Evolution.Methods Enzymol. 2018;608:59-79. doi: 10.1016/bs.mie.2018.04.006. Epub 2018 May 24. Methods Enzymol. 2018. PMID: 30173773
-
Polishing the craft of genetic diversity creation in directed evolution.Biotechnol Adv. 2013 Dec;31(8):1707-21. doi: 10.1016/j.biotechadv.2013.08.021. Epub 2013 Sep 6. Biotechnol Adv. 2013. PMID: 24012599 Review.
-
Directed evolution combined with synthetic biology strategies expedite semi-rational engineering of genes and genomes.Bioengineered. 2015;6(3):136-40. doi: 10.1080/21655979.2015.1011029. Epub 2015 Jan 26. Bioengineered. 2015. PMID: 25621864 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
