

A Novel COL4A5 Pathogenic Variant Joins the Dots in a Family with a Synchronous Diagnosis of Alport Syndrome and Polycystic Kidney Disease
- PMID: 38790225
- PMCID: PMC11121527
- DOI: 10.3390/genes15050597
A Novel COL4A5 Pathogenic Variant Joins the Dots in a Family with a Synchronous Diagnosis of Alport Syndrome and Polycystic Kidney Disease
Abstract
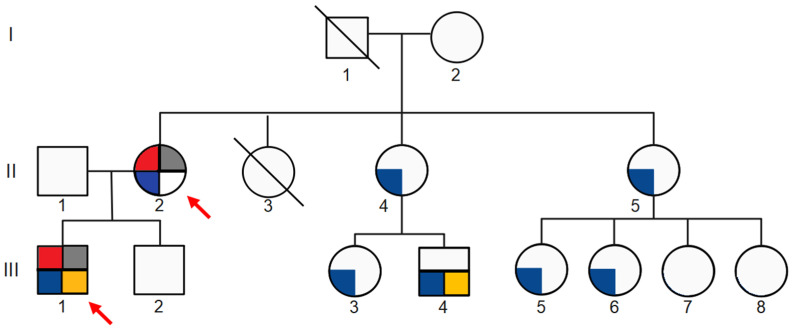
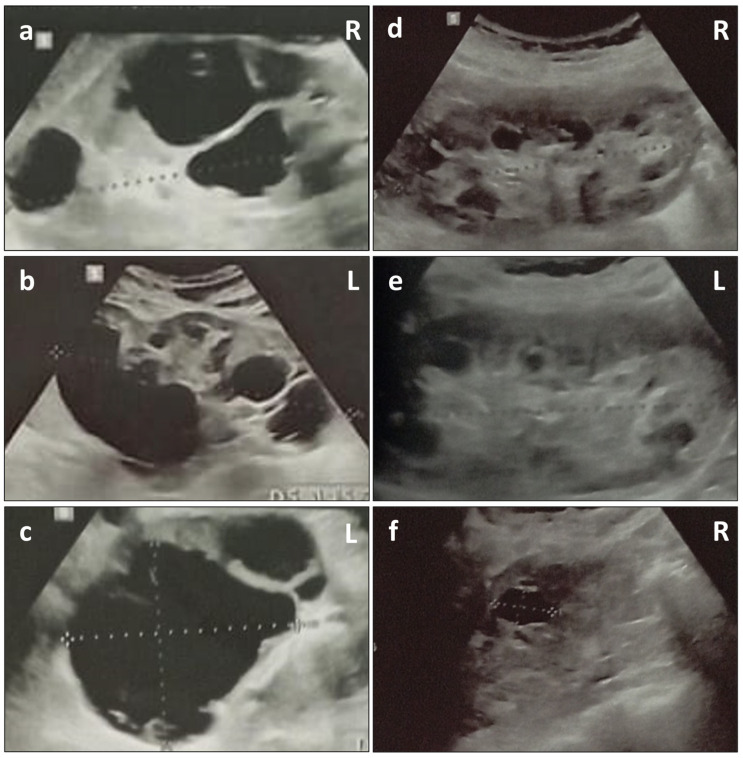
Alport Syndrome (AS) is the most common genetic glomerular disease, and it is caused by COL4A3, COL4A4, and COL4A5 pathogenic variants. The classic phenotypic spectrum associated with AS ranges from isolated hematuria to chronic kidney disease (CKD) with extrarenal abnormalities. Atypical presentation of the disorder is possible, and it can mislead the diagnosis. Polycystic kidney disease (PKD), which is most frequently associated with Autosomal Dominant PKD (ADPKD) due to PKD1 and PKD2 heterozygous variants, is emerging as a possible clinical manifestation in COL4A3-A5 patients. We describe a COL4A5 novel familial frameshift variant (NM_000495.5: c.1095dup p.(Leu366ValfsTer45)), which was associated with AS and PKD in the hemizygous proband, as well as with PKD, IgA glomerulonephritis and focal segmental glomerulosclerosis (FSGS) in the heterozygous mother. Establishing the diagnosis of AS can sometimes be difficult, especially in the context of misleading family history and atypical phenotypic features. This case study supports the emerging genotypic and phenotypic heterogeneity in COL4A3-A5-associated disorders, as well as the recently described association between PKD and collagen type IV (Col4) defects. We highlight the importance of the accurate phenotyping of all family members and the relevance of next-generation sequencing in the differential diagnosis of hereditary kidney disease.
Keywords: Alport Syndrome; COL4A5; phenotypic variability; polycystic kidney disease; sensorineural hearing loss.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Watson S., Padala S.A., Hashmi M.F., Bush J.S. StatPearls. StatPearls Publishing; Treasure Island, FL, USA: 2024. [(accessed on 21 April 2024)]. Alport Syndrome. Available online: http://www.ncbi.nlm.nih.gov/books/NBK470419/ - PubMed
-
- Savige J., Storey H., Watson E., Hertz J.M., Deltas C., Renieri A., Mari F., Hilbert P., Plevova P., Byers P., et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: Refining the ACMG criteria. Eur. J. Hum. Genet. 2021;29:1186–1197. doi: 10.1038/s41431-021-00858-1. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
