

BMPR2 Loss Activates AKT by Disrupting DLL4/NOTCH1 and PPARγ Signaling in Pulmonary Arterial Hypertension
- PMID: 38791441
- PMCID: PMC11121464
- DOI: 10.3390/ijms25105403
BMPR2 Loss Activates AKT by Disrupting DLL4/NOTCH1 and PPARγ Signaling in Pulmonary Arterial Hypertension
Abstract
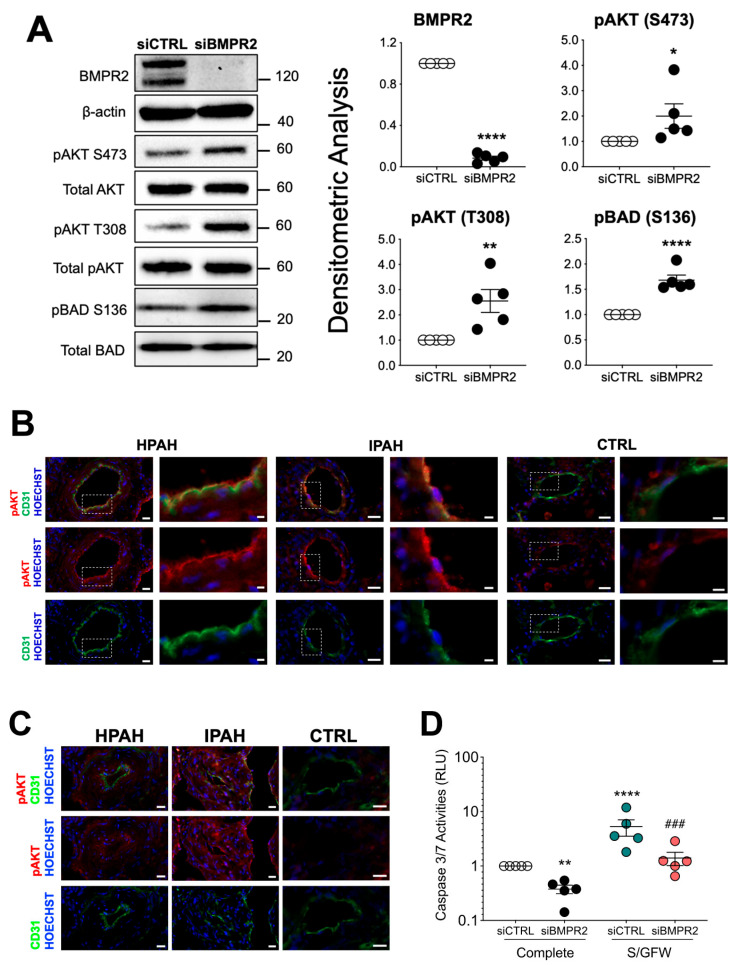
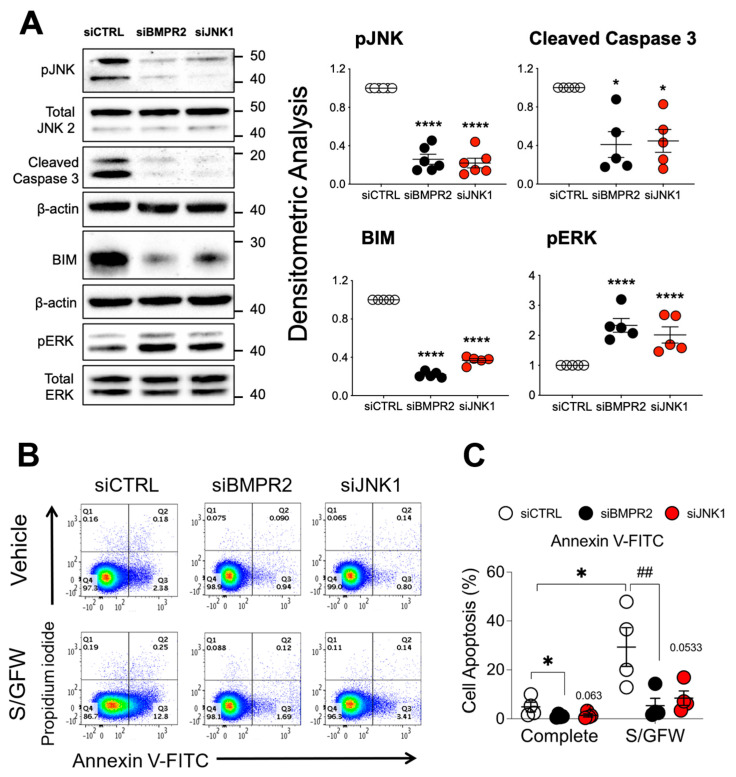
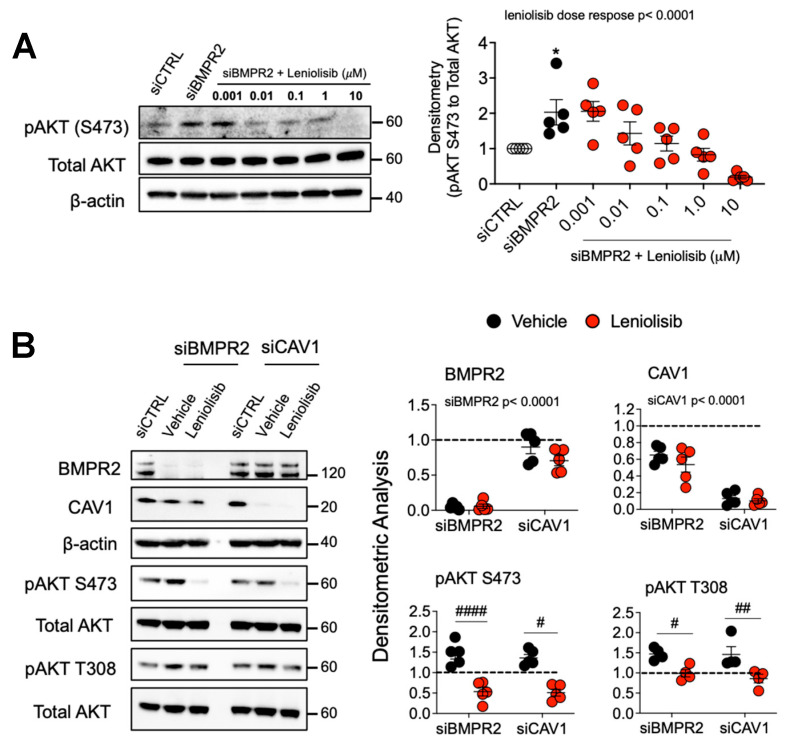
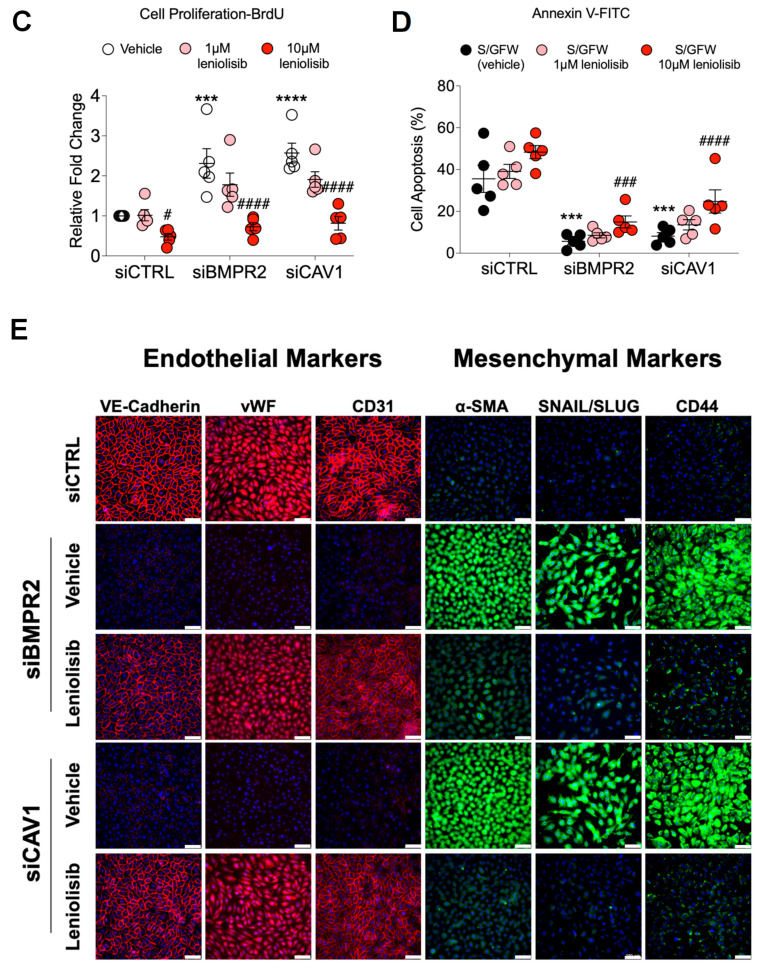
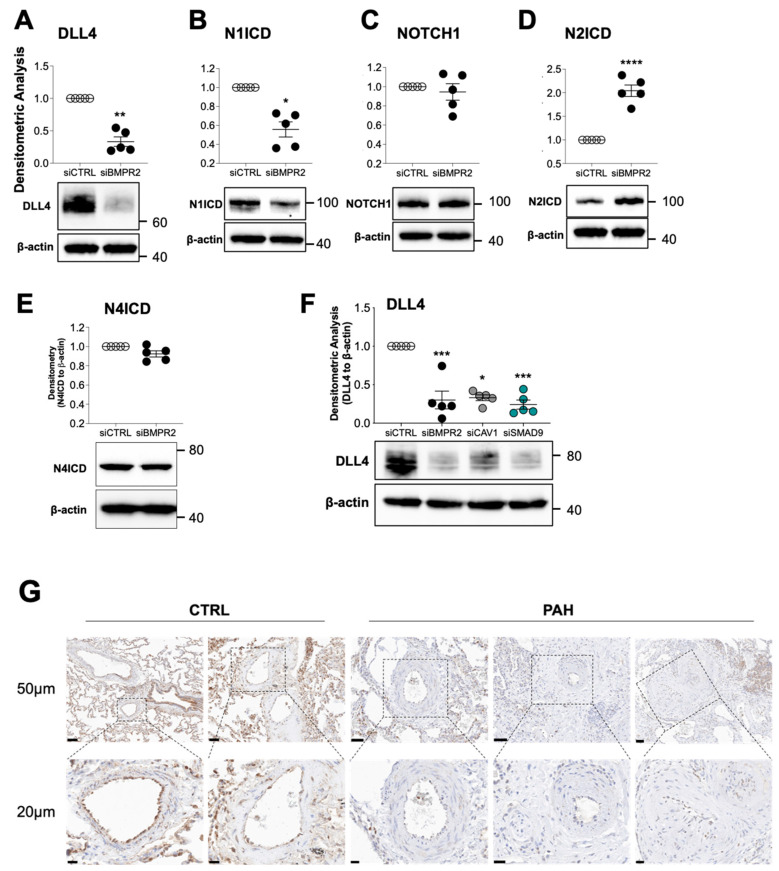
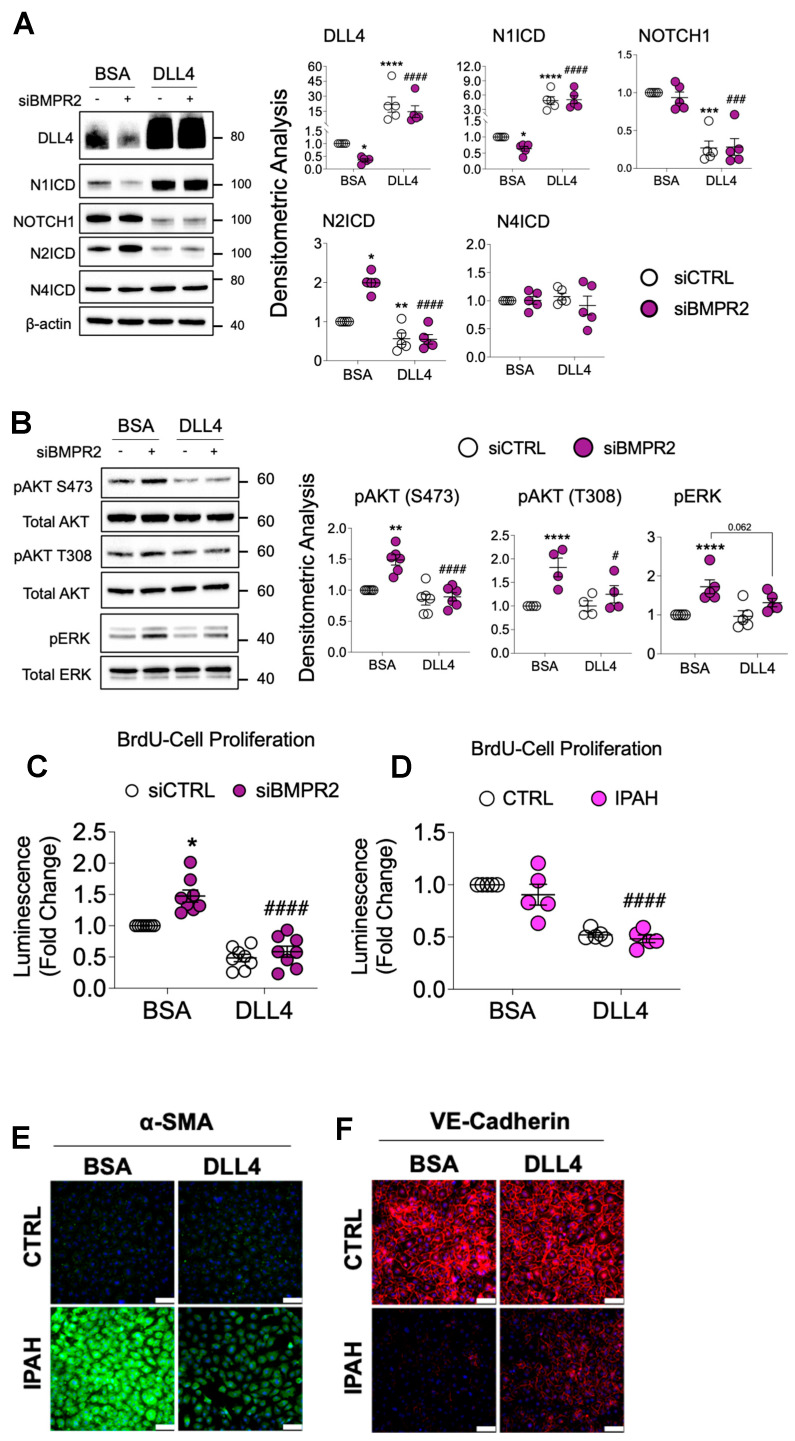
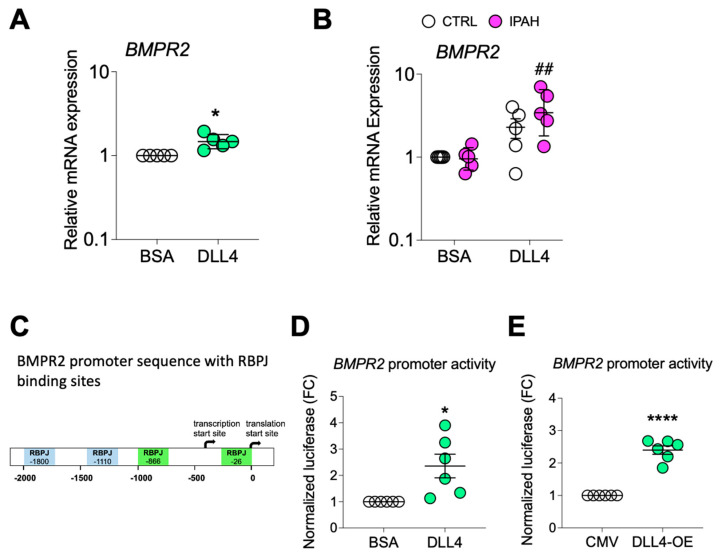
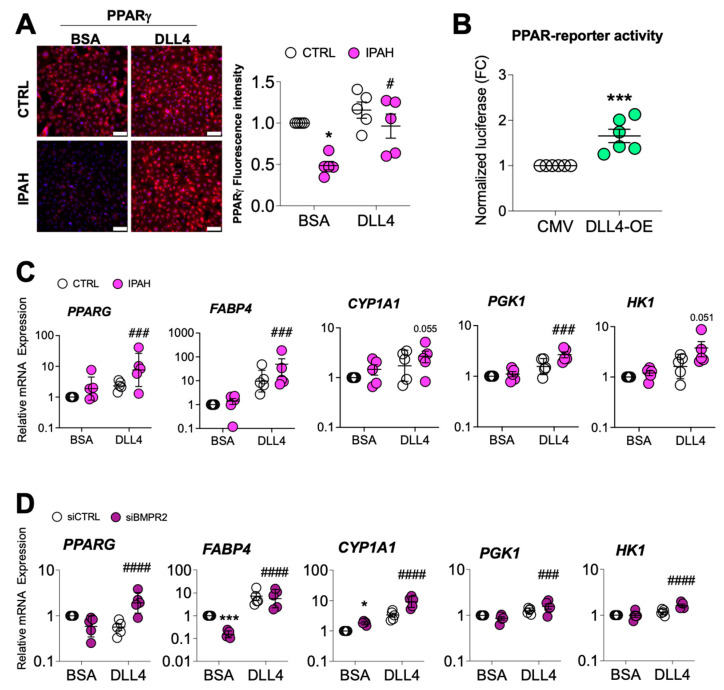
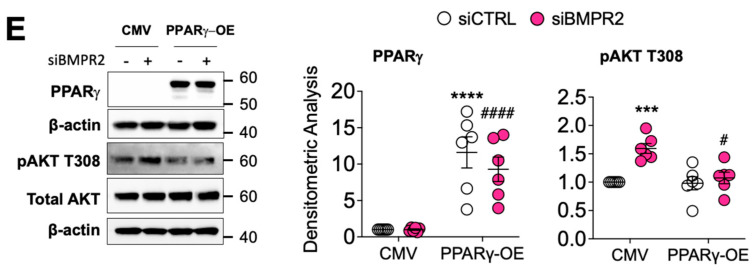
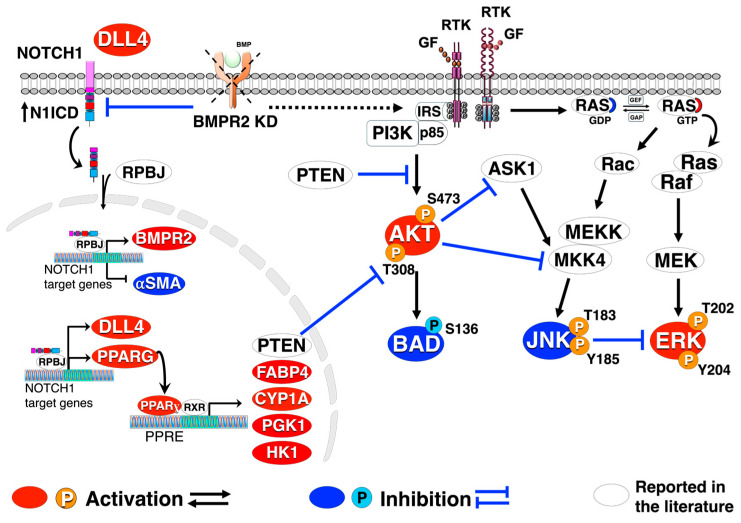
Pulmonary arterial hypertension (PAH) is a progressive cardiopulmonary disease characterized by pathologic vascular remodeling of small pulmonary arteries. Endothelial dysfunction in advanced PAH is associated with proliferation, apoptosis resistance, and endothelial to mesenchymal transition (EndoMT) due to aberrant signaling. DLL4, a cell membrane associated NOTCH ligand, plays a pivotal role maintaining vascular integrity. Inhibition of DLL4 has been associated with the development of pulmonary hypertension, but the mechanism is incompletely understood. Here we report that BMPR2 silencing in pulmonary artery endothelial cells (PAECs) activated AKT and suppressed the expression of DLL4. Consistent with these in vitro findings, increased AKT activation and reduced DLL4 expression was found in the small pulmonary arteries of patients with PAH. Increased NOTCH1 activation through exogenous DLL4 blocked AKT activation, decreased proliferation and reversed EndoMT. Exogenous and overexpression of DLL4 induced BMPR2 and PPRE promoter activity, and BMPR2 and PPARG mRNA in idiopathic PAH (IPAH) ECs. PPARγ, a nuclear receptor associated with EC homeostasis, suppressed by BMPR2 loss was induced and activated by DLL4/NOTCH1 signaling in both BMPR2-silenced and IPAH ECs, reversing aberrant phenotypic changes, in part through AKT inhibition. Directly blocking AKT or restoring DLL4/NOTCH1/PPARγ signaling may be beneficial in preventing or reversing the pathologic vascular remodeling of PAH.
Keywords: AKT; BMPR2; DLL4; PPARγ; endothelial dysfunction; pulmonary arterial hypertension; signal transduction; vascular remodeling.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Update of
-
Disruption of DLL4/NOTCH1 Causes Dysregulated PPARγ/AKT Signaling in Pulmonary Arterial Hypertension.bioRxiv [Preprint]. 2024 Feb 2:2024.01.31.578230. doi: 10.1101/2024.01.31.578230. bioRxiv. 2024. Update in: Int J Mol Sci. 2024 May 15;25(10):5403. doi: 10.3390/ijms25105403. PMID: 38903104 Free PMC article. Updated. Preprint.
References
-
- Tang H., Chen J., Fraidenburg D.R., Song S., Sysol J.R., Drennan A.R., Offermanns S., Ye R.D., Bonini M.G., Minshall R.D., et al. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2015;308:L208–L220. doi: 10.1152/ajplung.00242.2014. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
