

Downregulation of MYBL1 in endothelial cells contributes to atherosclerosis by repressing PLEKHM1-inducing autophagy
- PMID: 38797732
- PMCID: PMC11128406
- DOI: 10.1007/s10565-024-09873-6
Downregulation of MYBL1 in endothelial cells contributes to atherosclerosis by repressing PLEKHM1-inducing autophagy
Abstract
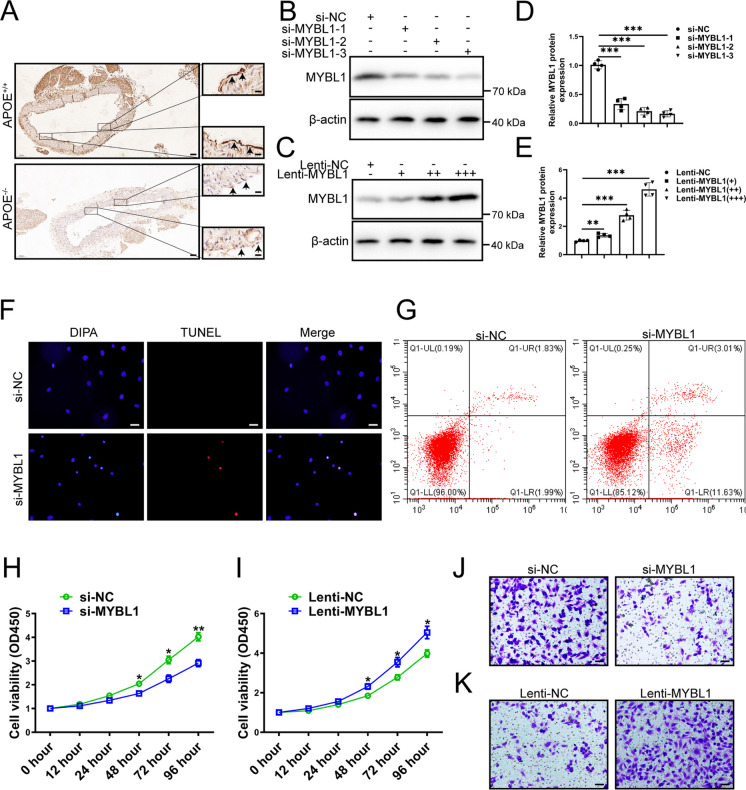
MYBL1 is a strong transcriptional activator involved in the cell signaling. However, there is no systematic study on the role of MYBL1 in atherosclerosis. The aim of this study is to elucidate the role and mechanism of MYBL1 in atherosclerosis. GSE28829, GSE43292 and GSE41571 were downloaded from NCBI for differentially expressed analysis. The expression levels of MYBL1 in atherosclerotic plaque tissue and normal vessels were detected by qRT-PCR, Western blot and Immunohistochemistry. Transwell and CCK-8 were used to detect the migration and proliferation of HUVECs after silencing MYBL1. RNA-seq, Western blot, qRT-PCR, Luciferase reporter system, Immunofluorescence, Flow cytometry, ChIP and CO-IP were used to study the role and mechanism of MYBL1 in atherosclerosis. The microarray data of GSE28829, GSE43292, and GSE41571 were analyzed and intersected, and then MYBL1 were verified. MYBL1 was down-regulated in atherosclerotic plaque tissue. After silencing of MYBL1, HUVECs were damaged, and their migration and proliferation abilities were weakened. Overexpression of MYBL1 significantly enhanced the migration and proliferation of HUVECs. MYBL1 knockdown induced abnormal autophagy in HUVEC cells, suggesting that MYBL1 was involved in the regulation of HUVECs through autophagy. Mechanistic studies showed that MYBL1 knockdown inhibited autophagosome and lysosomal fusion in HUVECs by inhibiting PLEKHM1, thereby exacerbating atherosclerosis. Furthermore, MYBL1 was found to repress lipid accumulation in HUVECs after oxLDL treatment. MYBL1 knockdown in HUVECs was involved in atherosclerosis by inhibiting PLEKHM1-induced autophagy, which provided a novel target of therapy for atherosclerosis.
Keywords: Atherosclerosis; Autophagy; MYB proto-oncogene-like 1; Pleckstrin Homology and RUN domain containing M1.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
The authors declare that they have no competing interests.
Figures








References
-
- Ayari H, Bricca G. Identification of two genes potentially associated in iron-heme homeostasis in human carotid plaque using microarray analysis. J Biosci. 2013;38(2):311–5. 10.1007/s12038-013-9310-2. - PubMed
-
- Chiu TH, Ku CW, Ho TJ, Tsai KL, Yang YD, Ou HC, et al. Schisanhenol ameliorates oxLDL-caused endothelial dysfunction by inhibiting LOX-1 signaling. Environ Toxicol. 2023;38(7):1589–96. 10.1002/tox.23788. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
