

This is a preprint.
A single-cell atlas characterizes dysregulation of the bone marrow immune microenvironment associated with outcomes in multiple myeloma
- PMID: 38798338
- PMCID: PMC11118283
- DOI: 10.1101/2024.05.15.593193
A single-cell atlas characterizes dysregulation of the bone marrow immune microenvironment associated with outcomes in multiple myeloma
Update in
-
A single-cell atlas characterizes dysregulation of the bone marrow immune microenvironment associated with outcomes in multiple myeloma.Nat Cancer. 2026 Jan;7(1):224-246. doi: 10.1038/s43018-025-01072-4. Epub 2026 Jan 9. Nat Cancer. 2026. PMID: 41514053 Free PMC article.
Abstract
Multiple Myeloma (MM) remains incurable despite advances in treatment options. Although tumor subtypes and specific DNA abnormalities are linked to worse prognosis, the impact of immune dysfunction on disease emergence and/or treatment sensitivity remains unclear. We established a harmonized consortium to generate an Immune Atlas of MM aimed at informing disease etiology, risk stratification, and potential therapeutic strategies. We generated a transcriptome profile of 1,149,344 single cells from the bone marrow of 263 newly diagnosed patients enrolled in the CoMMpass study and characterized immune and hematopoietic cell populations. Associating cell abundances and gene expression with disease progression revealed the presence of a proinflammatory immune senescence-associated secretory phenotype in rapidly progressing patients. Furthermore, signaling analyses suggested active intercellular communication involving APRIL-BCMA, potentially promoting tumor growth and survival. Finally, we demonstrate that integrating immune cell levels with genetic information can significantly improve patient stratification.
Keywords: Bone Marrow Microenvironment; Inflammation; Multiple Myeloma; Senescence; Single-Cell; Transcriptome.
Figures

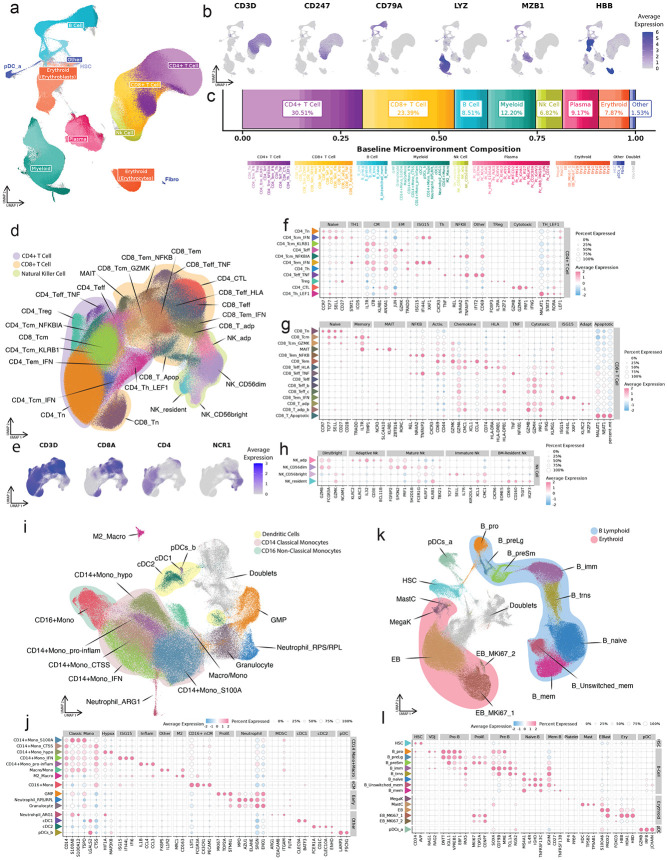





References
-
- Mikhael J., Bhutani M. & Cole C. E. Multiple myeloma for the primary care provider: A practical review to promote earlier diagnosis among diverse populations. Am. J. Med. 136, 33–41 (2023). - PubMed
-
- Cancer Stat Facts: Myeloma. National Cancer Institute Surveillance, Epidemiology, and End Results Program https://seer.cancer.gov/statfacts/html/mulmy.html.
-
- Avet-Loiseau H. et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood 109, 3489–3495 (2007). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous