

This is a preprint.
Sex-specific single cell-level transcriptomic signatures of Rett syndrome disease progression
- PMID: 38798575
- PMCID: PMC11118571
- DOI: 10.1101/2024.05.16.594595
Sex-specific single cell-level transcriptomic signatures of Rett syndrome disease progression
Update in
-
Sex-specific single cell-level transcriptomic signatures of Rett syndrome disease progression.Commun Biol. 2024 Oct 10;7(1):1292. doi: 10.1038/s42003-024-06990-0. Commun Biol. 2024. PMID: 39384967 Free PMC article.
Abstract
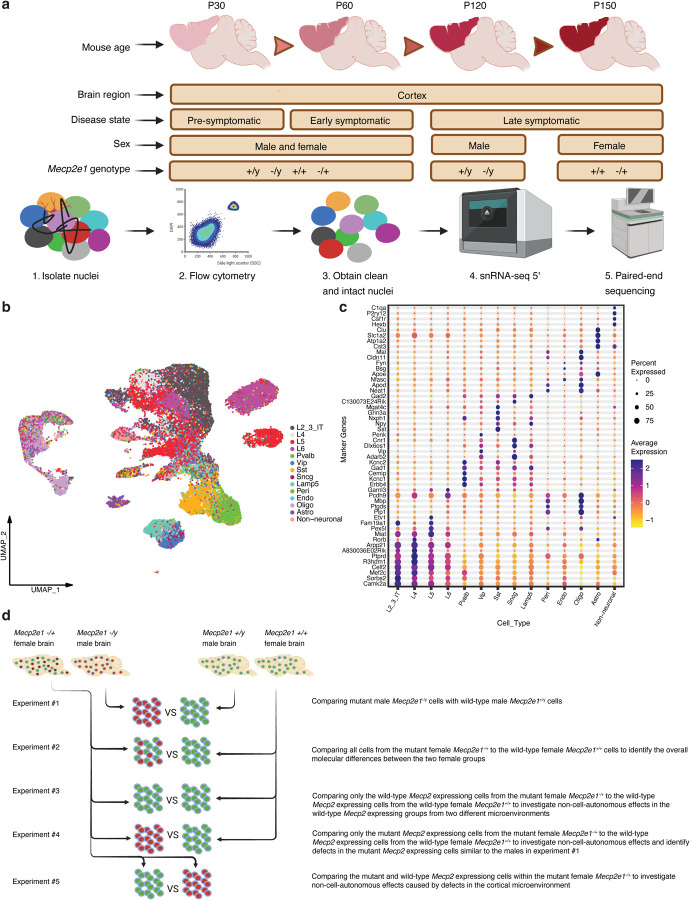
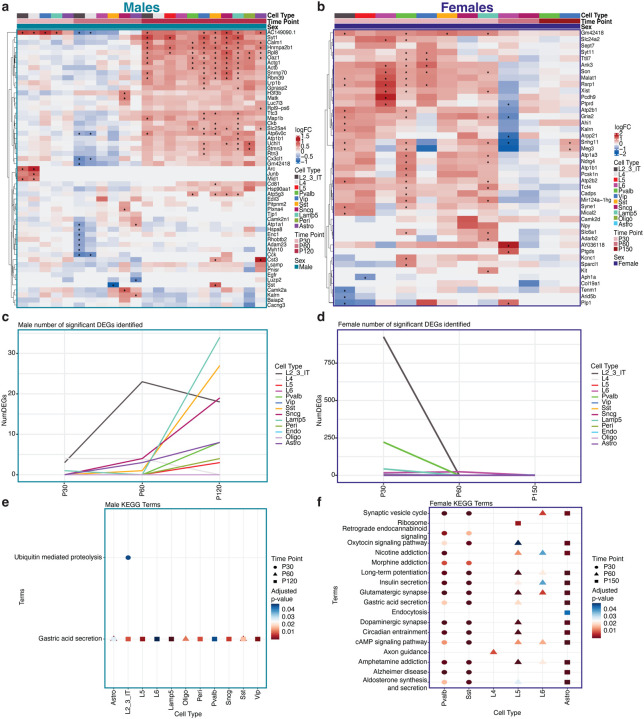
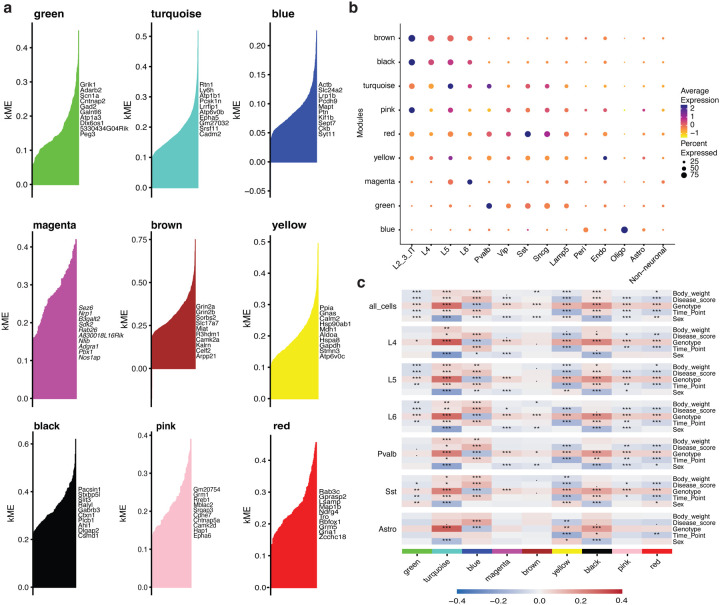
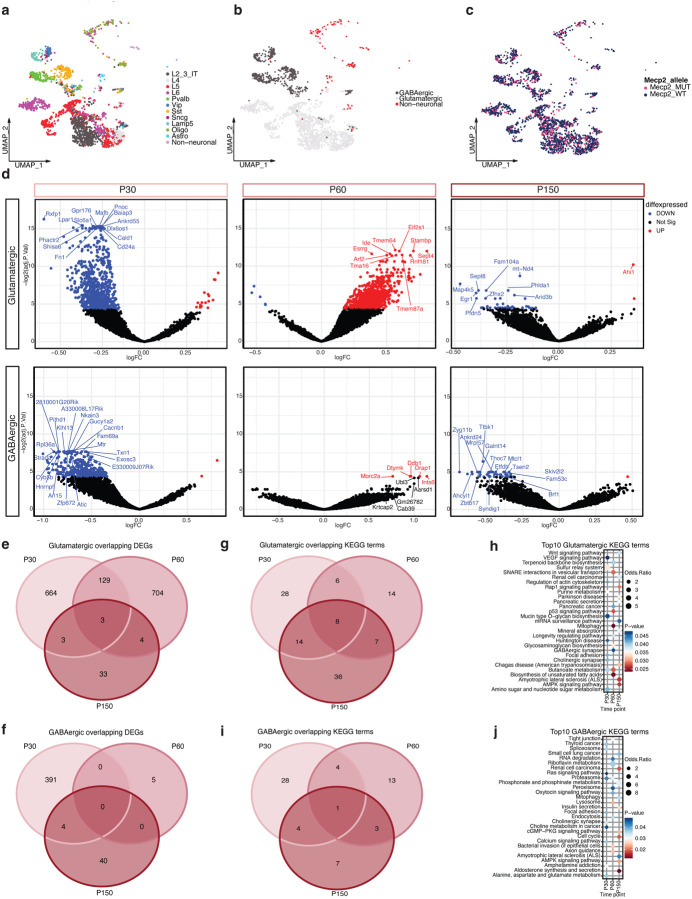
Dominant X-linked diseases are uncommon due to female X chromosome inactivation (XCI). While random XCI usually protects females against X-linked mutations, Rett syndrome (RTT) is a female neurodevelopmental disorder caused by heterozygous MECP2 mutation. After 6-18 months of typical neurodevelopment, RTT girls undergo poorly understood regression. We performed longitudinal snRNA-seq on cerebral cortex in a construct-relevant Mecp2e1 mutant mouse model of RTT, revealing transcriptional effects of cell type, mosaicism, and sex on progressive disease phenotypes. Across cell types, we observed sex differences in the number of differentially expressed genes (DEGs) with 6x more DEGs in mutant females than males. Unlike males, female DEGs emerged prior to symptoms, were enriched for homeostatic gene pathways in distinct cell types over time, and correlated with disease phenotypes and human RTT cortical cell transcriptomes. Non-cell-autonomous effects were prominent and dynamic across disease progression of Mecp2e1 mutant females, indicating wild-type-expressing cells normalizing transcriptional homeostasis. These results improve understanding of RTT progression and treatment.
Figures
References
-
- Amir R. E. et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat. Genet. 23, 185–188 (1999). - PubMed
-
- Zoghbi H. Y. MeCP2 dysfunction in humans and mice. J. Child Neurol. 20, 736–740 (2005). - PubMed
-
- Guy J., Hendrich B., Holmes M., Martin J. E. & Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic rett syndrome. Nat. Genet. 27, 322–326 (2001). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources