

Native dynamics and allosteric responses in PTP1B probed by high-resolution HDX-MS
- PMID: 38801229
- PMCID: PMC11129624
- DOI: 10.1002/pro.5024
Native dynamics and allosteric responses in PTP1B probed by high-resolution HDX-MS
Abstract
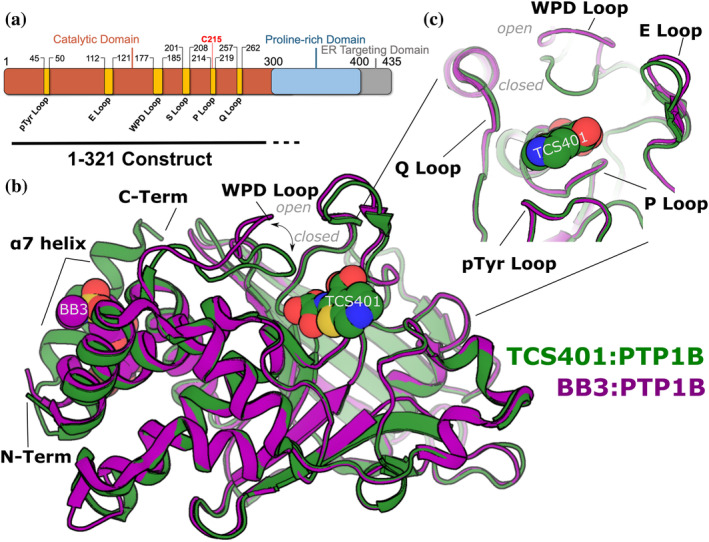
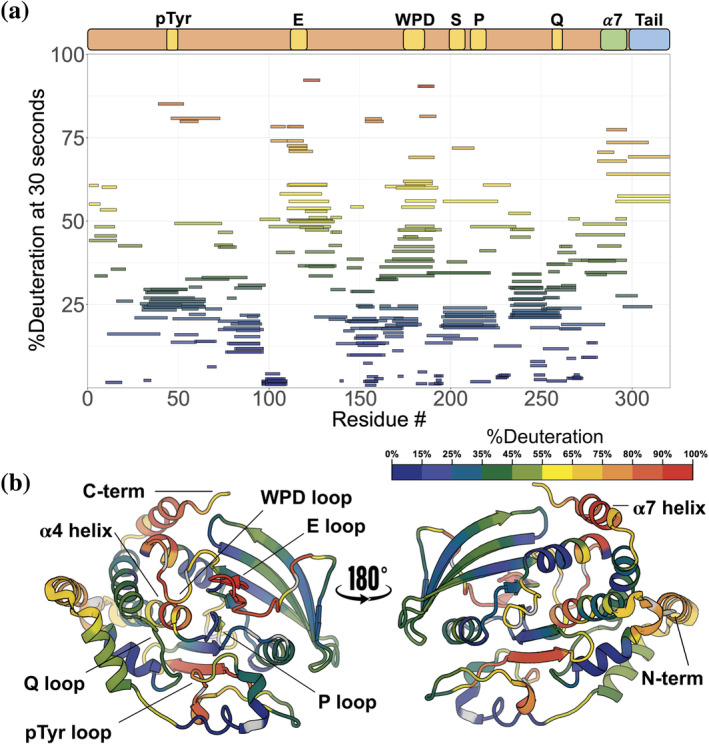
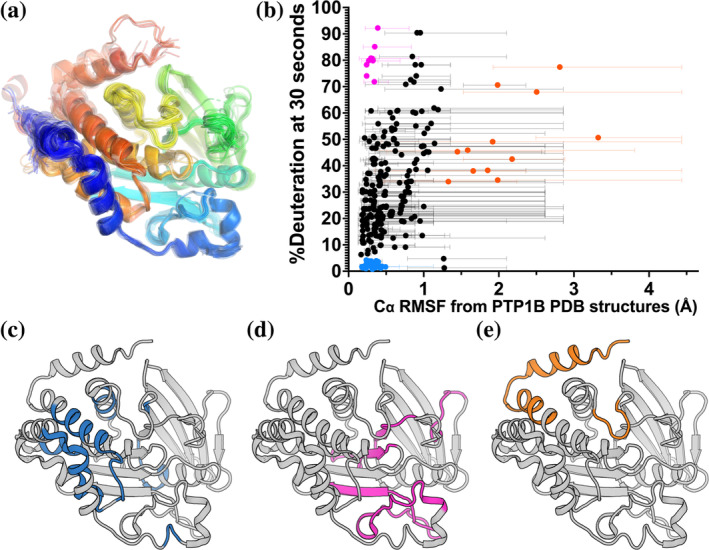
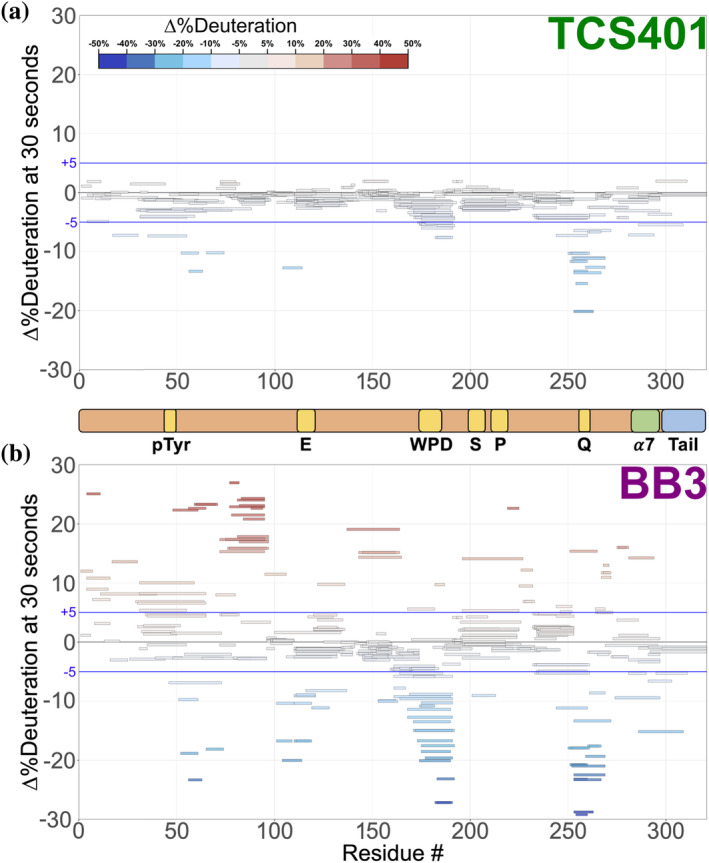
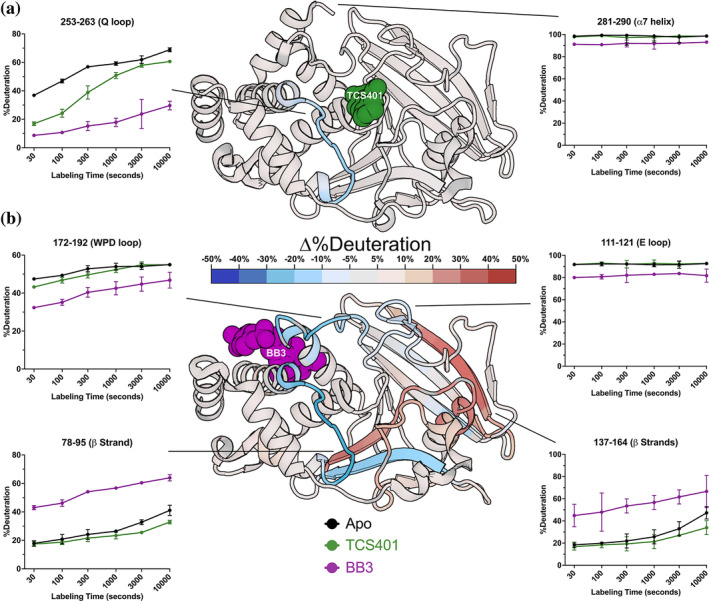
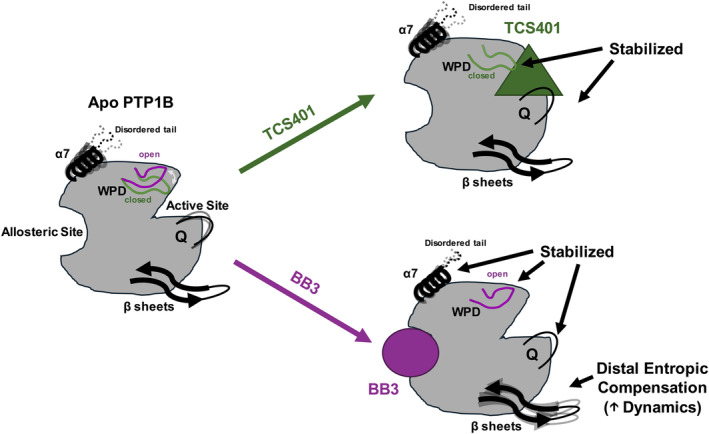
Protein tyrosine phosphatase 1B (PTP1B) is a validated therapeutic target for obesity, diabetes, and certain types of cancer. In particular, allosteric inhibitors hold potential for therapeutic use, but an incomplete understanding of conformational dynamics and allostery in this protein has hindered their development. Here, we interrogate solution dynamics and allosteric responses in PTP1B using high-resolution hydrogen-deuterium exchange mass spectrometry (HDX-MS), an emerging and powerful biophysical technique. Using HDX-MS, we obtain a detailed map of backbone amide exchange that serves as a proxy for the solution dynamics of apo PTP1B, revealing several flexible loops interspersed among more constrained and rigid regions within the protein structure, as well as local regions that exchange faster than expected from their secondary structure and solvent accessibility. We demonstrate that our HDX rate data obtained in solution adds value to estimates of conformational heterogeneity derived from a pseudo-ensemble constructed from ~200 crystal structures of PTP1B. Furthermore, we report HDX-MS maps for PTP1B with active-site versus allosteric small-molecule inhibitors. These maps suggest distinct and widespread effects on protein dynamics relative to the apo form, including changes in locations distal (>35 Å) from the respective ligand binding sites. These results illuminate that allosteric inhibitors of PTP1B can induce unexpected changes in dynamics that extend beyond the previously understood allosteric network. Together, our data suggest a model of BB3 allostery in PTP1B that combines conformational restriction of active-site residues with compensatory liberation of distal residues that aid in entropic balancing. Overall, our work showcases the potential of HDX-MS for elucidating aspects of protein conformational dynamics and allosteric effects of small-molecule ligands and highlights the potential of integrating HDX-MS alongside other complementary methods, such as room-temperature X-ray crystallography, NMR spectroscopy, and molecular dynamics simulations, to guide the development of new therapeutics.
Keywords: HDX‐MS; X‐ray crystallography; allostery; mass spectrometry; protein dynamics; protein structure; structural bioinformatics.
© 2024 The Authors. Protein Science published by Wiley Periodicals LLC on behalf of The Protein Society.
Figures
Update of
-
Native dynamics and allosteric responses in PTP1B probed by high-resolution HDX-MS.bioRxiv [Preprint]. 2023 Nov 13:2023.07.12.548582. doi: 10.1101/2023.07.12.548582. bioRxiv. 2023. Update in: Protein Sci. 2024 Jun;33(6):e5024. doi: 10.1002/pro.5024. PMID: 37503000 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
