

Ebselen analogues delay disease onset and its course in fALS by on-target SOD-1 engagement
- PMID: 38802492
- PMCID: PMC11130262
- DOI: 10.1038/s41598-024-62903-5
Ebselen analogues delay disease onset and its course in fALS by on-target SOD-1 engagement
Abstract
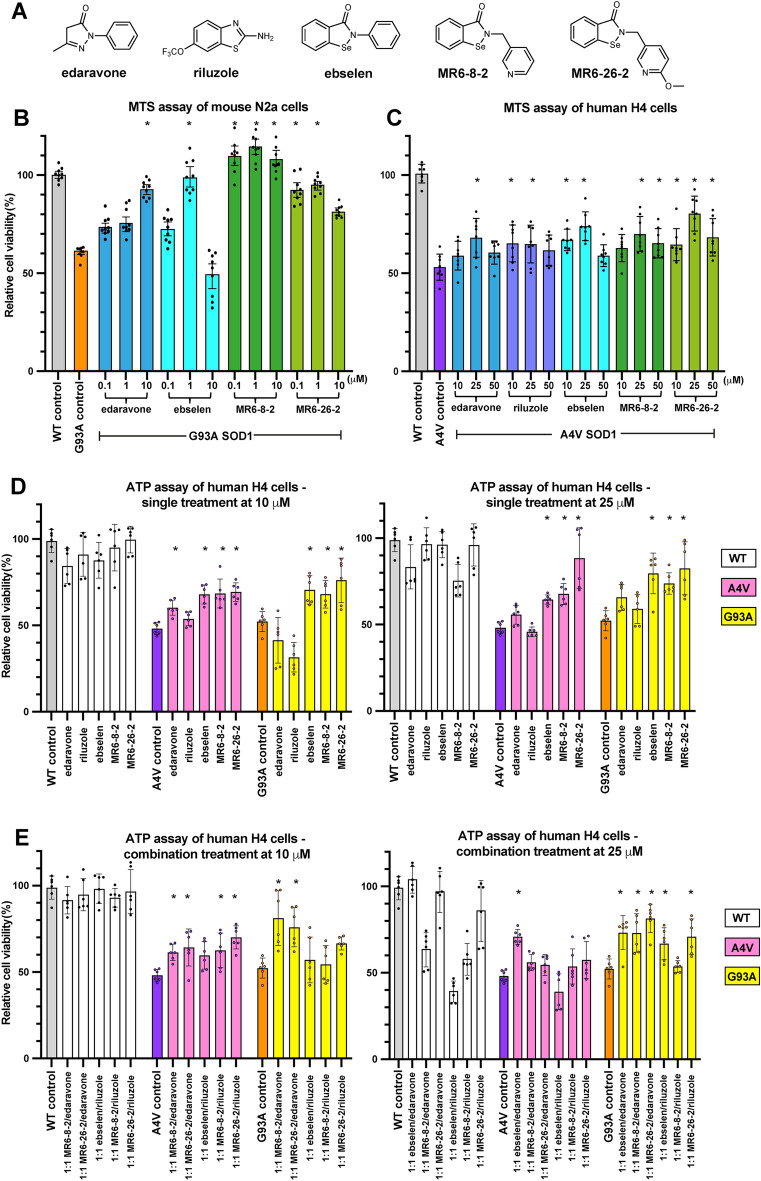
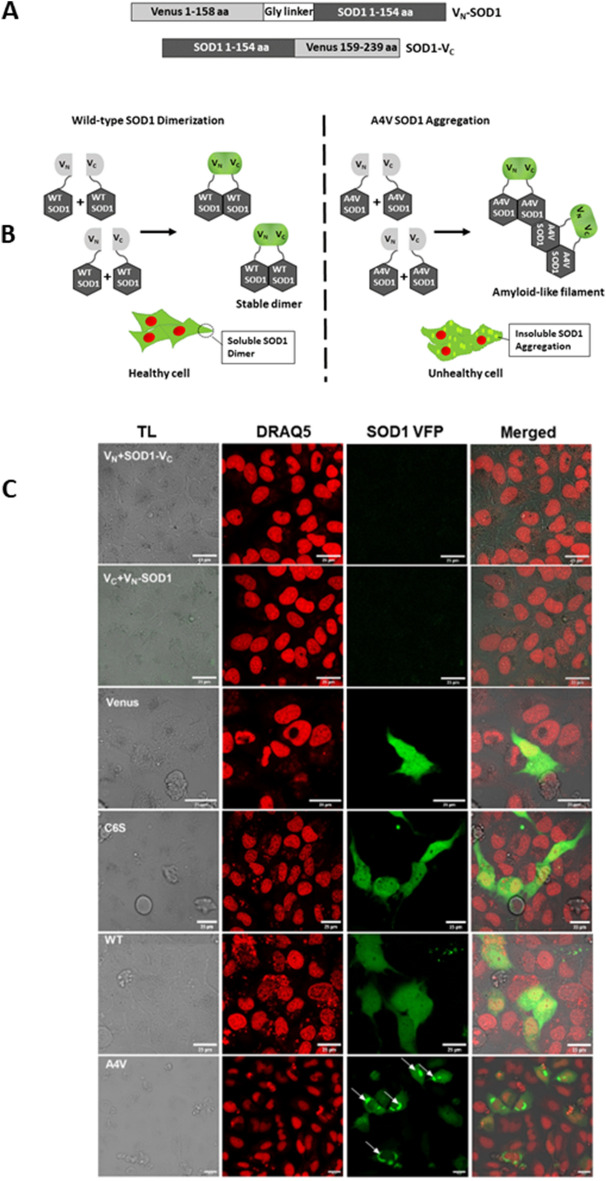
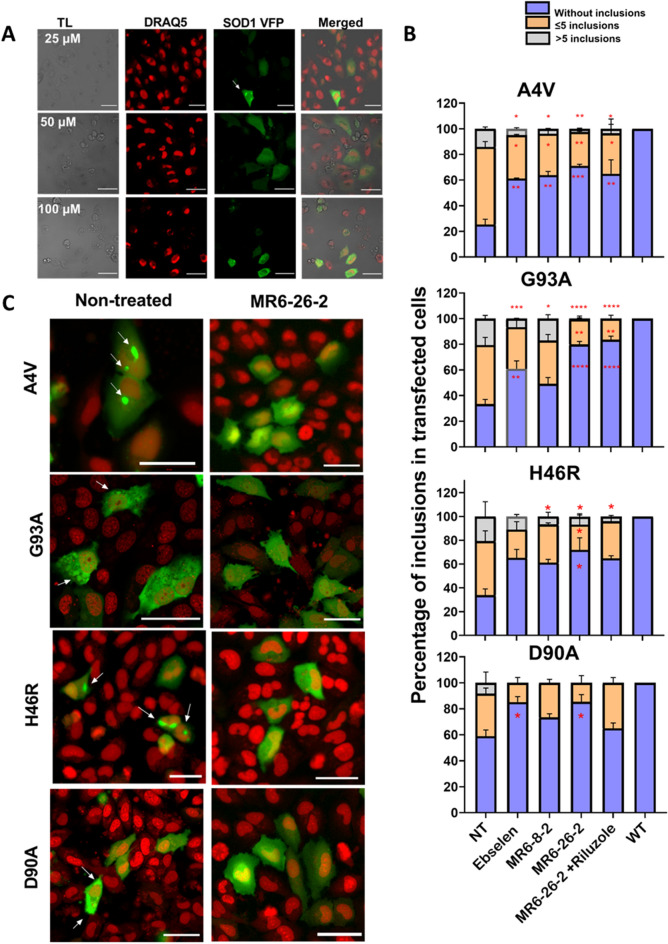
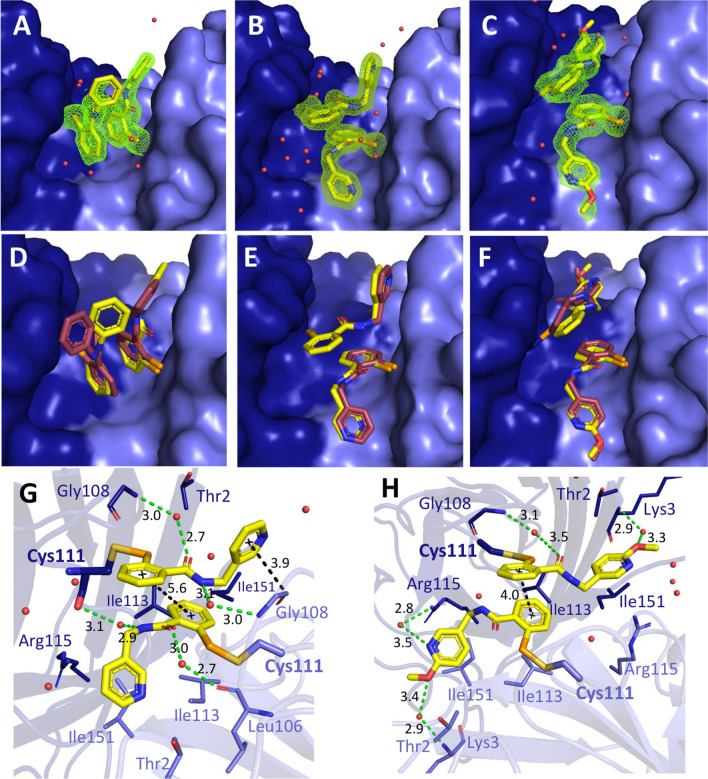
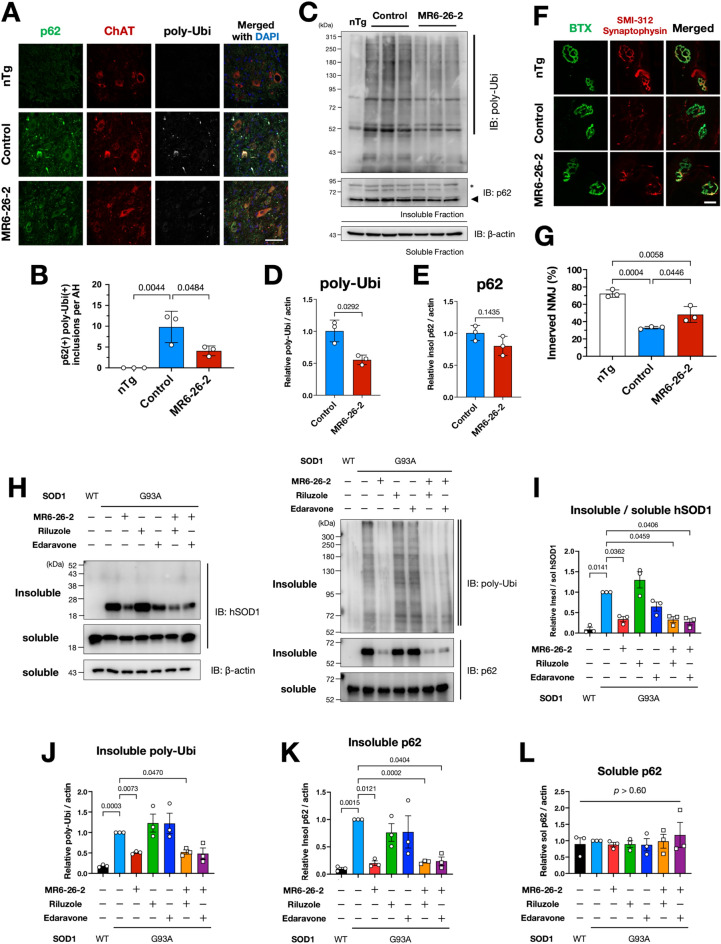
Amyotrophic lateral sclerosis (ALS) selectively affects motor neurons. SOD1 is the first causative gene to be identified for ALS and accounts for at least 20% of the familial (fALS) and up to 4% of sporadic (sALS) cases globally with some geographical variability. The destabilisation of the SOD1 dimer is a key driving force in fALS and sALS. Protein aggregation resulting from the destabilised SOD1 is arrested by the clinical drug ebselen and its analogues (MR6-8-2 and MR6-26-2) by redeeming the stability of the SOD1 dimer. The in vitro target engagement of these compounds is demonstrated using the bimolecular fluorescence complementation assay with protein-ligand binding directly visualised by co-crystallography in G93A SOD1. MR6-26-2 offers neuroprotection slowing disease onset of SOD1G93A mice by approximately 15 days. It also protected neuromuscular junction from muscle denervation in SOD1G93A mice clearly indicating functional improvement.
Keywords: Amyotrophic lateral sclerosis; Drug development; Ebselen; Motor neuron disease; Riluzole; Superoxide dismutase; Target engagement.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
