

IGHMBP2 deletion suppresses translation and activates the integrated stress response
- PMID: 38803225
- PMCID: PMC11109757
- DOI: 10.26508/lsa.202302554
IGHMBP2 deletion suppresses translation and activates the integrated stress response
Abstract
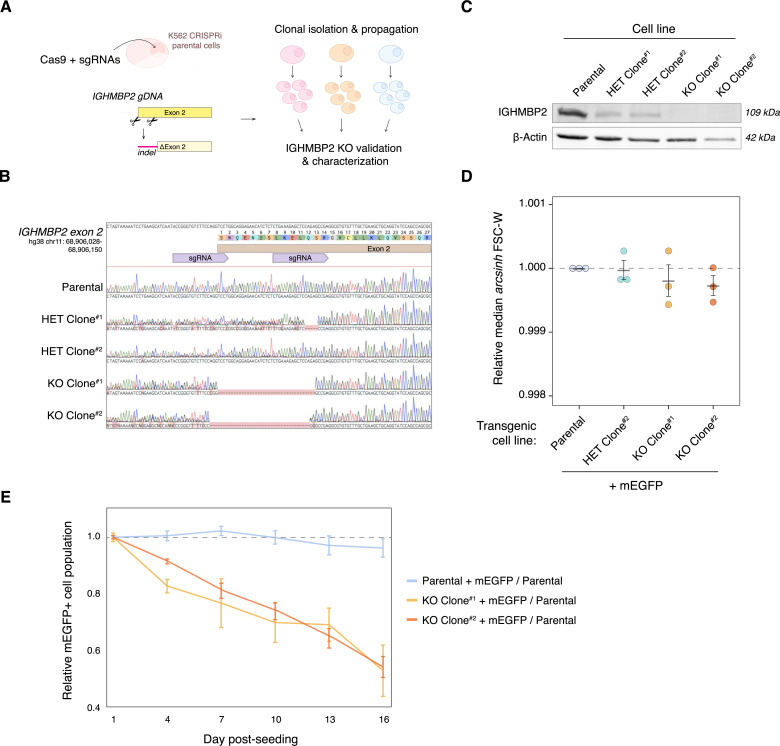
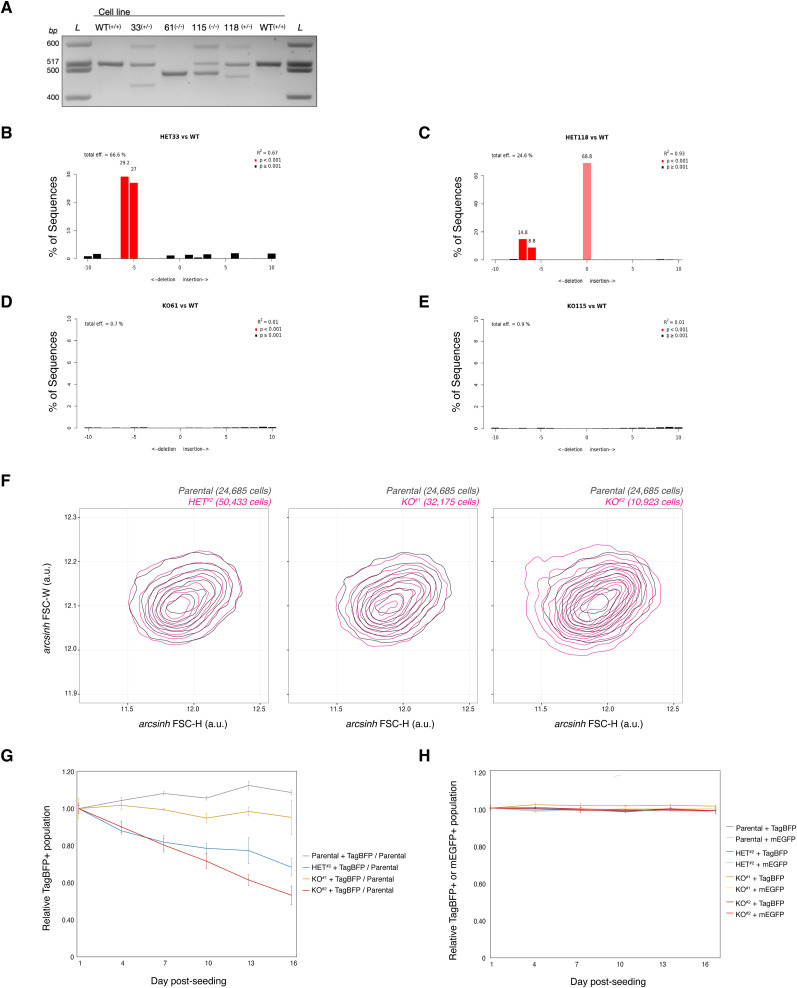
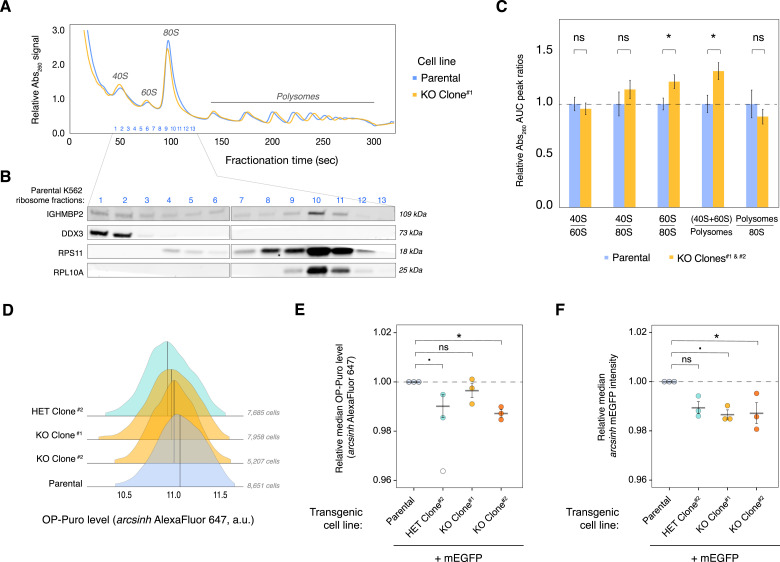
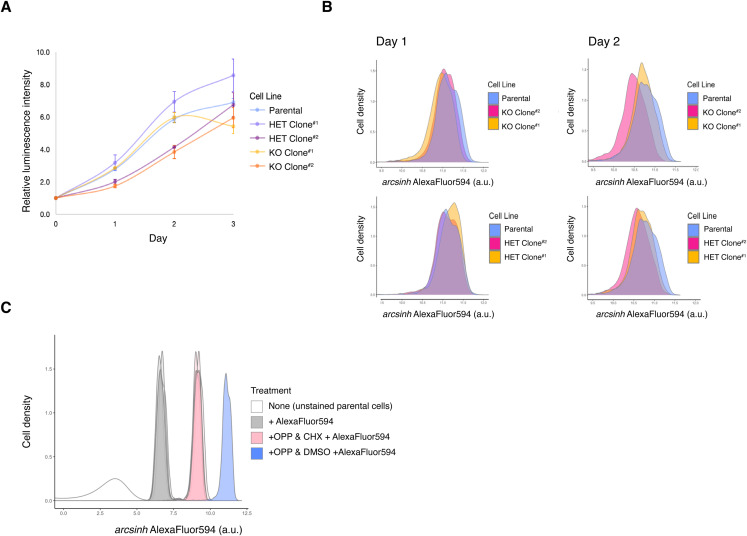
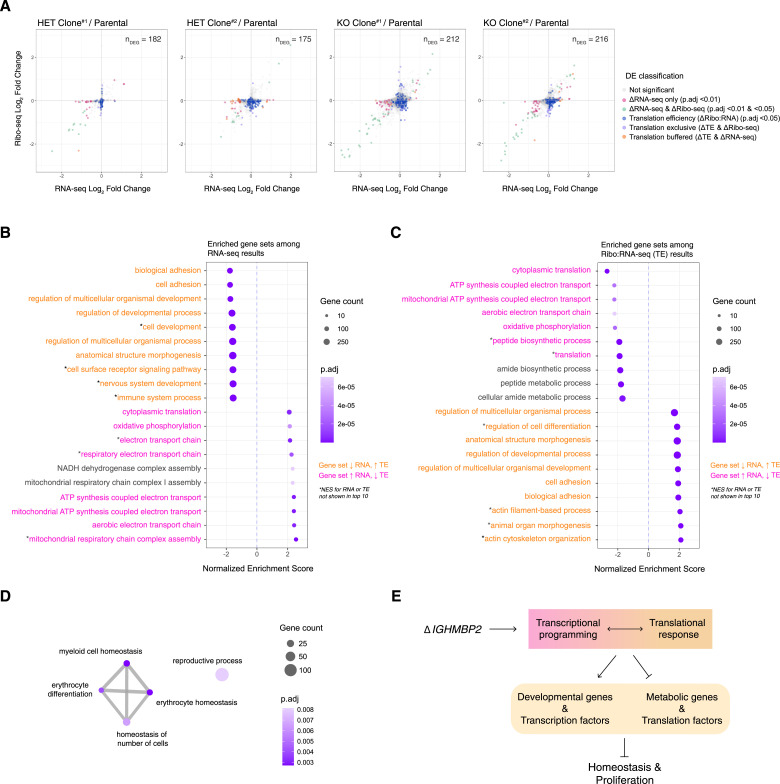
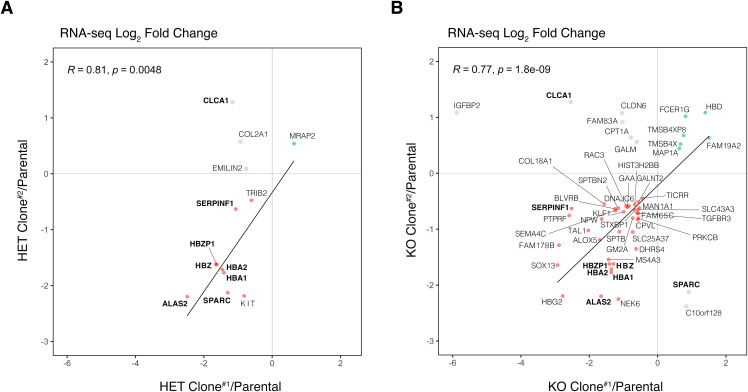
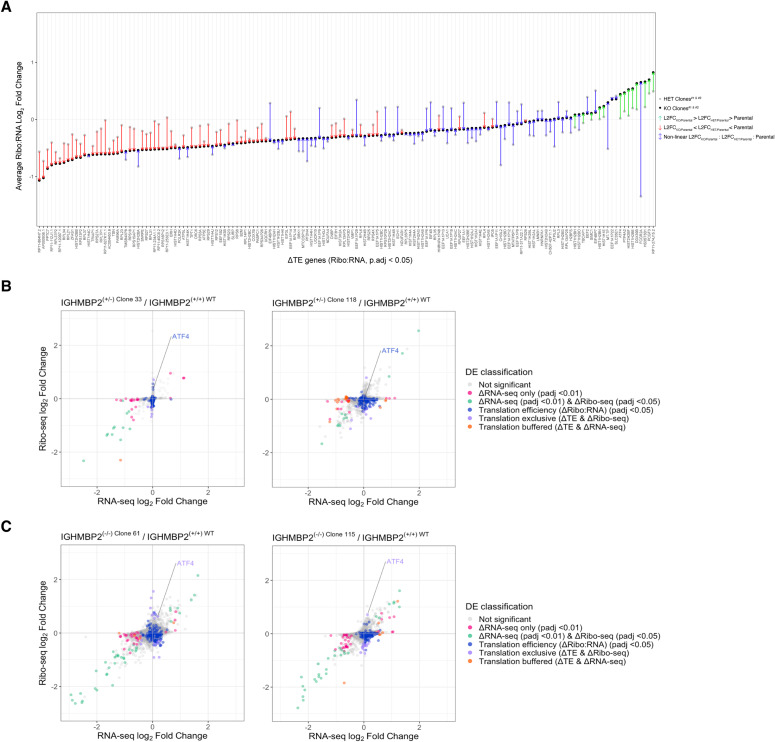
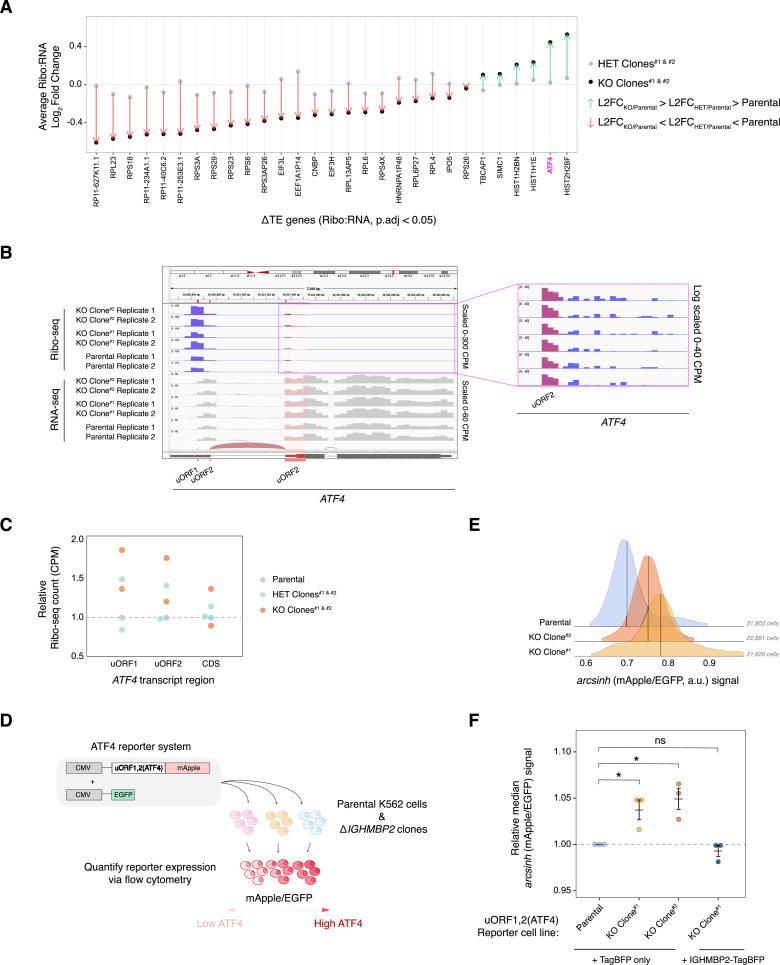
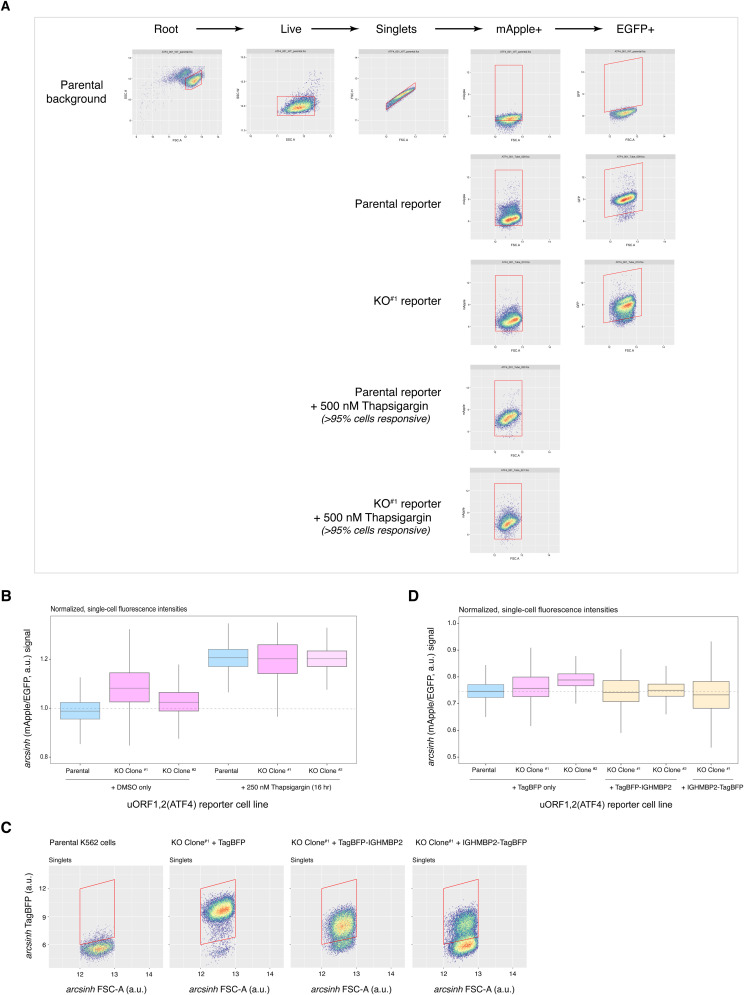
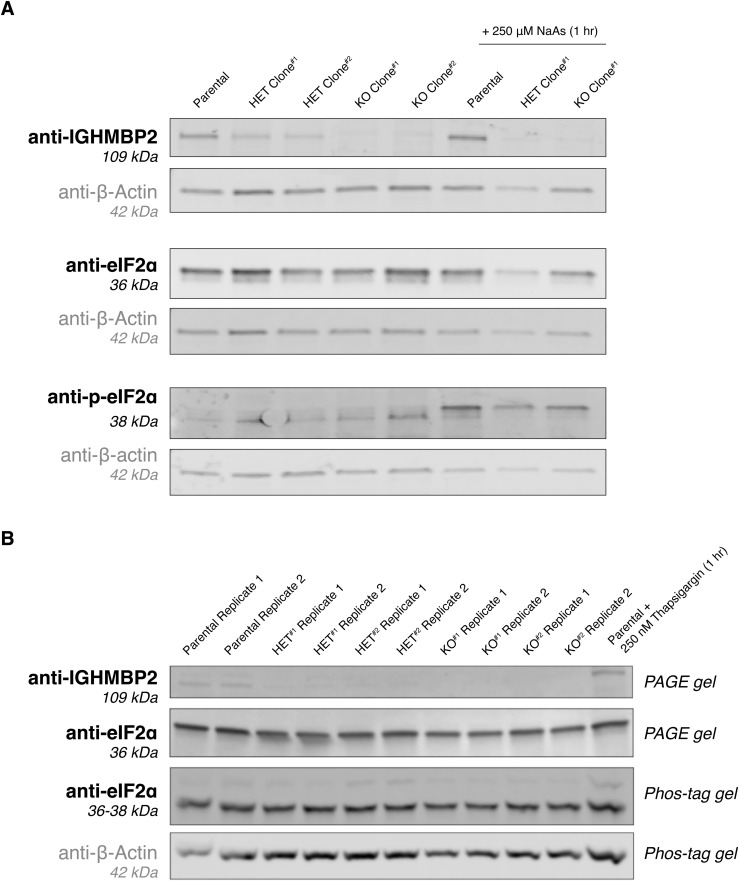
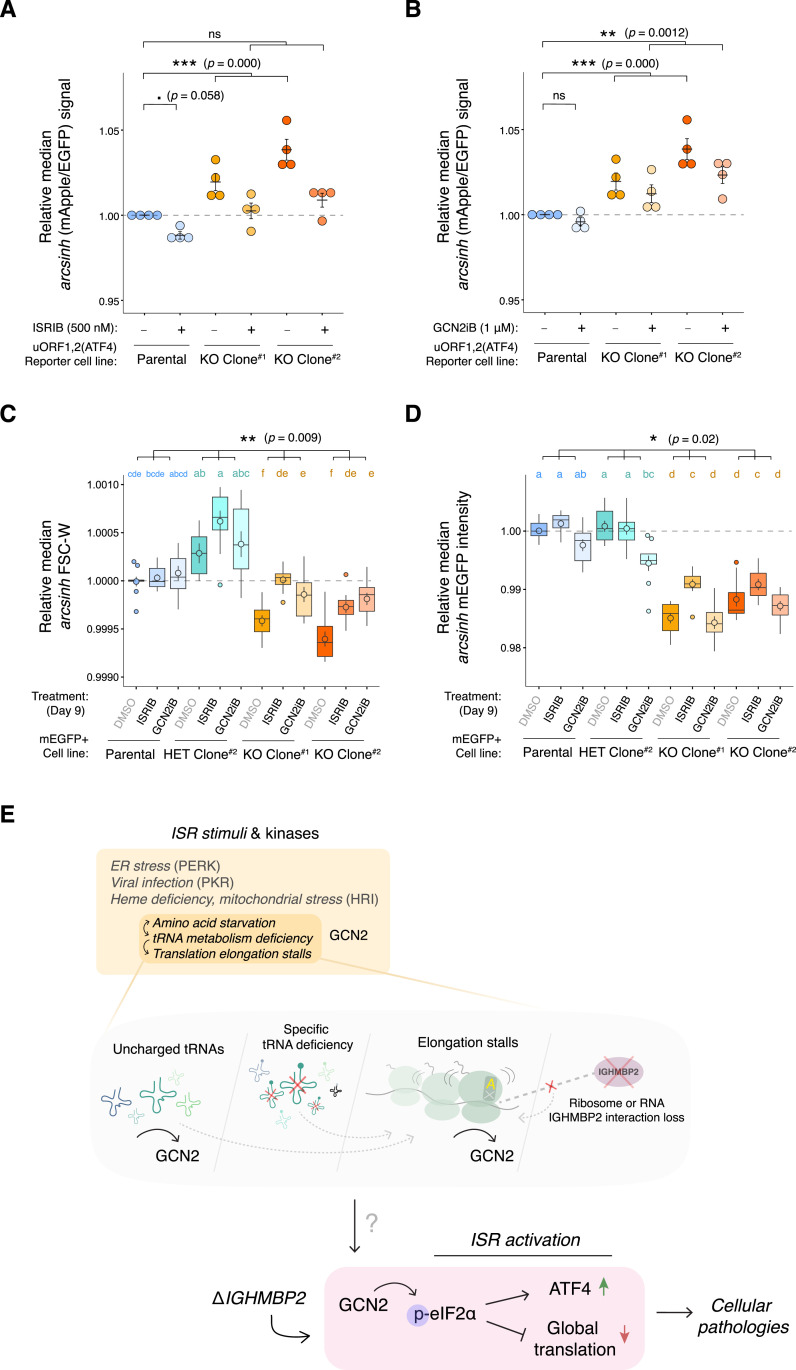
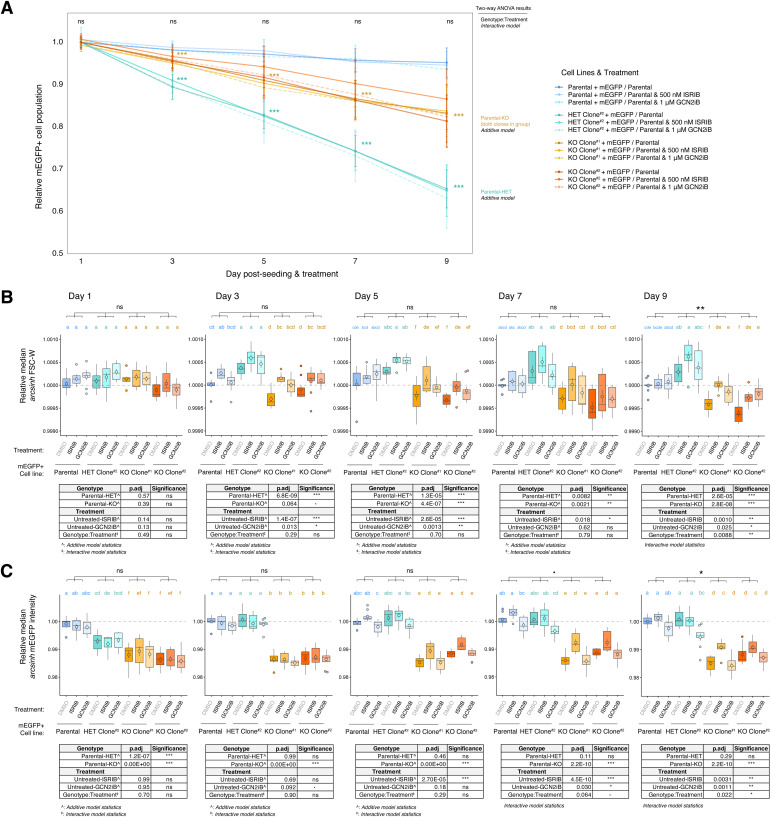
IGHMBP2 is a nonessential, superfamily 1 DNA/RNA helicase that is mutated in patients with rare neuromuscular diseases SMARD1 and CMT2S. IGHMBP2 is implicated in translational and transcriptional regulation via biochemical association with ribosomal proteins, pre-rRNA processing factors, and tRNA-related species. To uncover the cellular consequences of perturbing IGHMBP2, we generated full and partial IGHMBP2 deletion K562 cell lines. Using polysome profiling and a nascent protein synthesis assay, we found that IGHMBP2 deletion modestly reduces global translation. We performed Ribo-seq and RNA-seq and identified diverse gene expression changes due to IGHMBP2 deletion, including ATF4 up-regulation. With recent studies showing the integrated stress response (ISR) can contribute to tRNA metabolism-linked neuropathies, we asked whether perturbing IGHMBP2 promotes ISR activation. We generated ATF4 reporter cell lines and found IGHMBP2 knockout cells demonstrate basal, chronic ISR activation. Our work expands upon the impact of IGHMBP2 in translation and elucidates molecular mechanisms that may link mutant IGHMBP2 to severe clinical phenotypes.
© 2024 Park et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
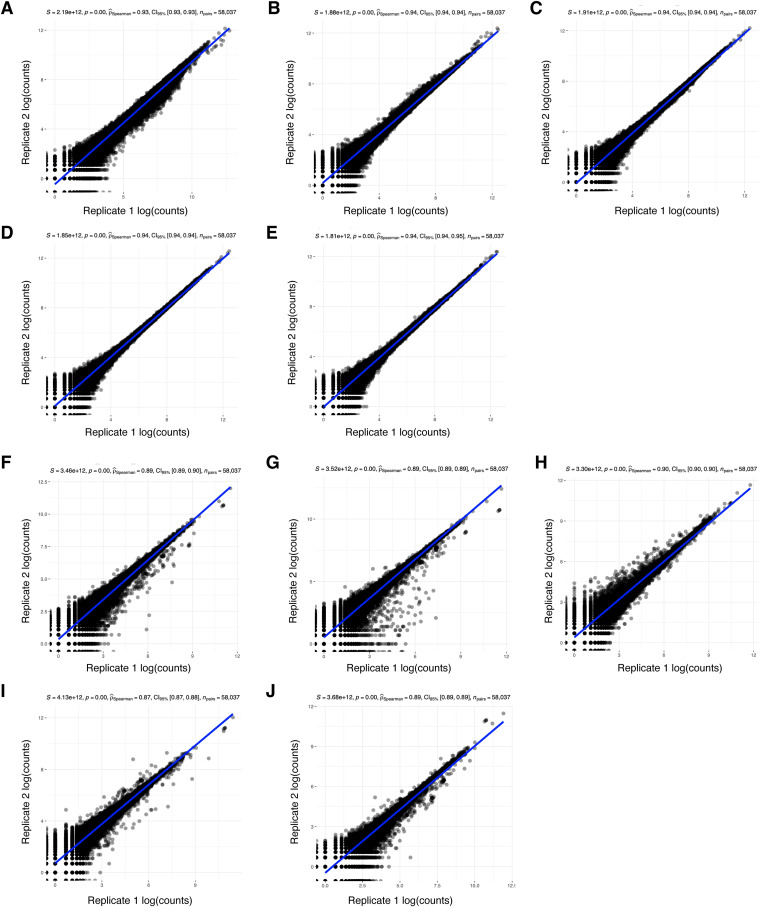
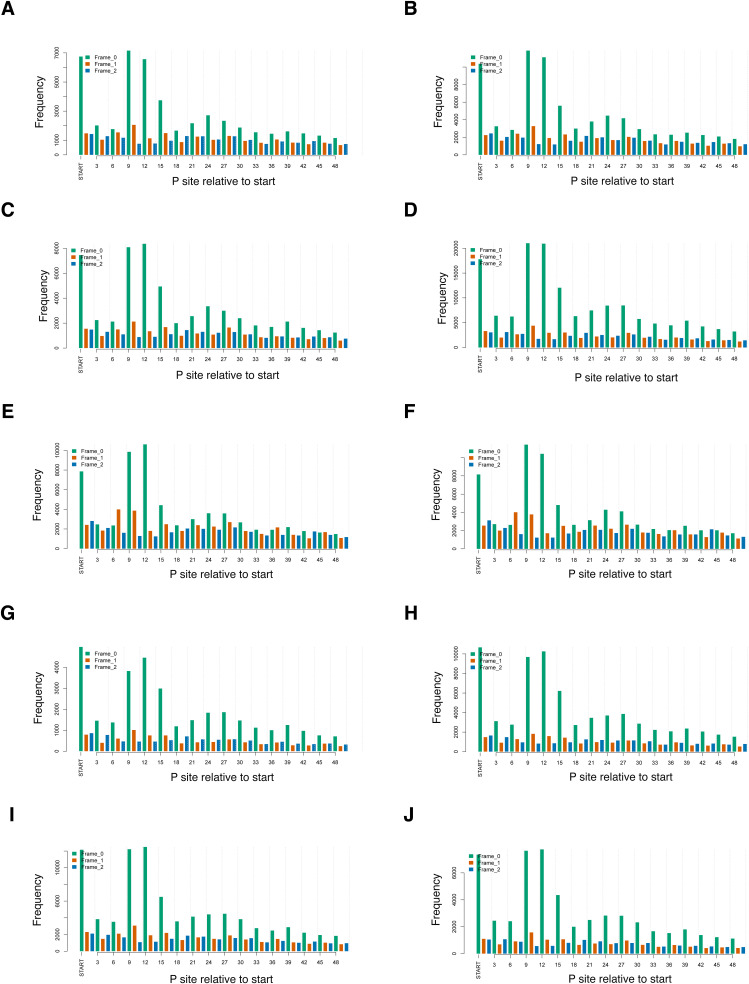
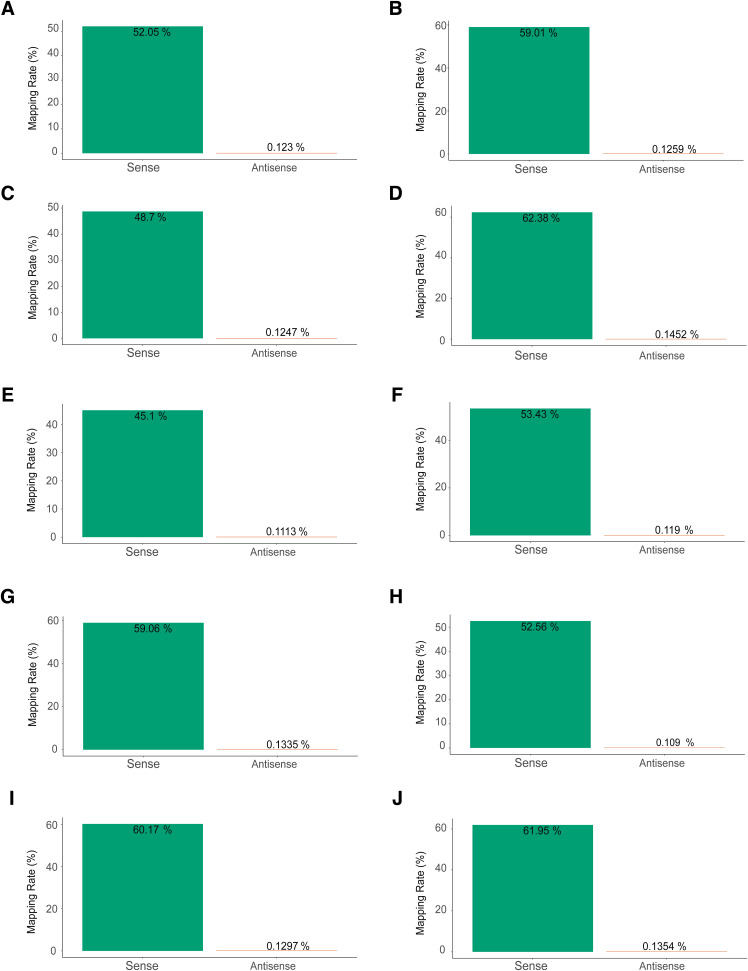
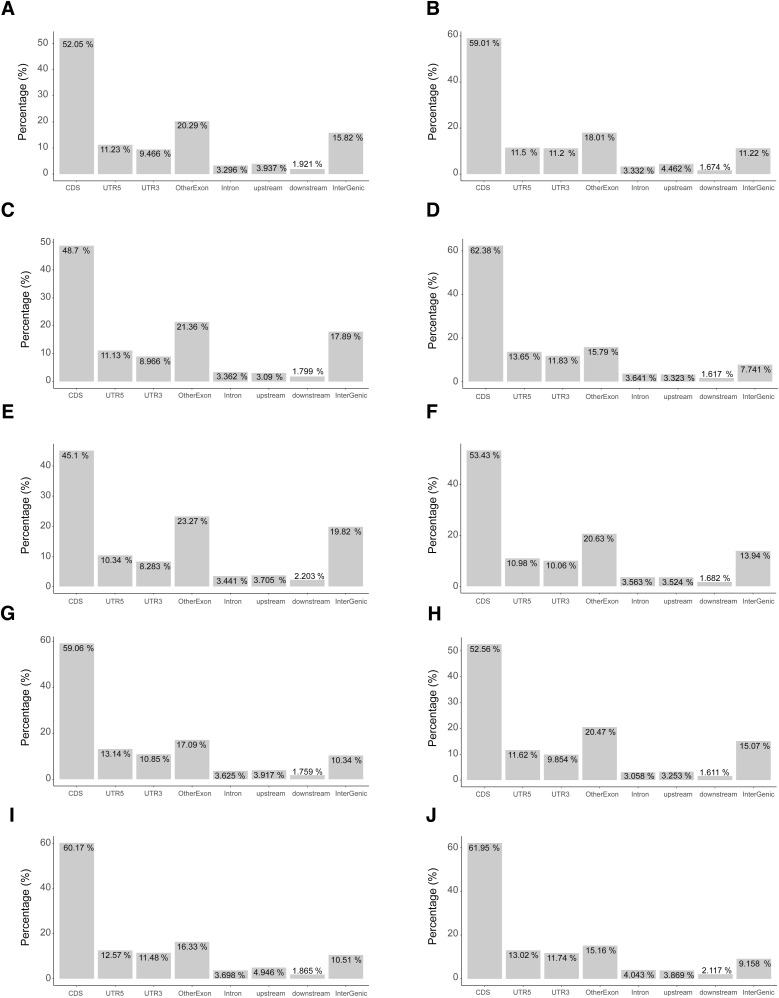
Update of
-
IGHMBP2 deletion suppresses translation and activates the integrated stress response.bioRxiv [Preprint]. 2023 Dec 12:2023.12.11.571166. doi: 10.1101/2023.12.11.571166. bioRxiv. 2023. Update in: Life Sci Alliance. 2024 May 21;7(8):e202302554. doi: 10.26508/lsa.202302554. PMID: 38168189 Free PMC article. Updated. Preprint.
Similar articles
-
IGHMBP2 deletion suppresses translation and activates the integrated stress response.bioRxiv [Preprint]. 2023 Dec 12:2023.12.11.571166. doi: 10.1101/2023.12.11.571166. bioRxiv. 2023. Update in: Life Sci Alliance. 2024 May 21;7(8):e202302554. doi: 10.26508/lsa.202302554. PMID: 38168189 Free PMC article. Updated. Preprint.
-
Impaired Local Translation of β-actin mRNA in Ighmbp2-Deficient Motoneurons: Implications for Spinal Muscular Atrophy with respiratory Distress (SMARD1).Neuroscience. 2018 Aug 21;386:24-40. doi: 10.1016/j.neuroscience.2018.06.019. Epub 2018 Jun 19. Neuroscience. 2018. PMID: 29928949
-
ABT1 modifies SMARD1 pathology via interactions with IGHMBP2 and stimulation of ATPase and helicase activity.JCI Insight. 2023 Jan 24;8(2):e164608. doi: 10.1172/jci.insight.164608. JCI Insight. 2023. PMID: 36480289 Free PMC article.
-
The molecular mechanisms that underlie IGHMBP2-related diseases.Neuropathol Appl Neurobiol. 2024 Aug;50(4):e13005. doi: 10.1111/nan.13005. Neuropathol Appl Neurobiol. 2024. PMID: 39119929 Review.
-
Models for IGHMBP2-associated diseases: an overview and a roadmap for the future.Neuromuscul Disord. 2021 Dec;31(12):1266-1278. doi: 10.1016/j.nmd.2021.08.001. Epub 2021 Aug 13. Neuromuscul Disord. 2021. PMID: 34785121 Review.
Cited by
-
Current Treatment Methods for Charcot-Marie-Tooth Diseases.Biomolecules. 2024 Sep 9;14(9):1138. doi: 10.3390/biom14091138. Biomolecules. 2024. PMID: 39334903 Free PMC article. Review.
-
Context-specific inhibition of mitochondrial ribosomes by phenicol and oxazolidinone antibiotics.bioRxiv [Preprint]. 2024 Aug 21:2024.08.21.609012. doi: 10.1101/2024.08.21.609012. bioRxiv. 2024. Update in: Nucleic Acids Res. 2025 Jan 24;53(3):gkaf046. doi: 10.1093/nar/gkaf046. PMID: 39229136 Free PMC article. Updated. Preprint.
-
Context-specific inhibition of mitochondrial ribosomes by phenicol and oxazolidinone antibiotics.Nucleic Acids Res. 2025 Jan 24;53(3):gkaf046. doi: 10.1093/nar/gkaf046. Nucleic Acids Res. 2025. PMID: 39907106 Free PMC article.
-
Neuroaxonal Degeneration as a Converging Mechanism in Motor Neuron Diseases (MNDs): Molecular Insights into RNA Dysregulation and Emerging Therapeutic Targets.Int J Mol Sci. 2025 Aug 7;26(15):7644. doi: 10.3390/ijms26157644. Int J Mol Sci. 2025. PMID: 40806770 Free PMC article. Review.
-
Integrated multi-omics analysis of zinc-finger proteins uncovers roles in RNA regulation.Mol Cell. 2024 Oct 3;84(19):3826-3842.e8. doi: 10.1016/j.molcel.2024.08.010. Epub 2024 Sep 19. Mol Cell. 2024. PMID: 39303722 Free PMC article.
References
-
- Calviello L, Sydow D, Harnett D, Ohler U (2019) Ribo-seQC: Comprehensive analysis of cytoplasmic and organellar ribosome profiling data. BioRxiv. 10.1101/601468 (Preprint posted April 08, 2019). - DOI