

Utilization of automated cilia analysis to characterize novel INPP5E variants in patients with non-syndromic retinitis pigmentosa
- PMID: 38806661
- PMCID: PMC11576733
- DOI: 10.1038/s41431-024-01627-6
Utilization of automated cilia analysis to characterize novel INPP5E variants in patients with non-syndromic retinitis pigmentosa
Abstract
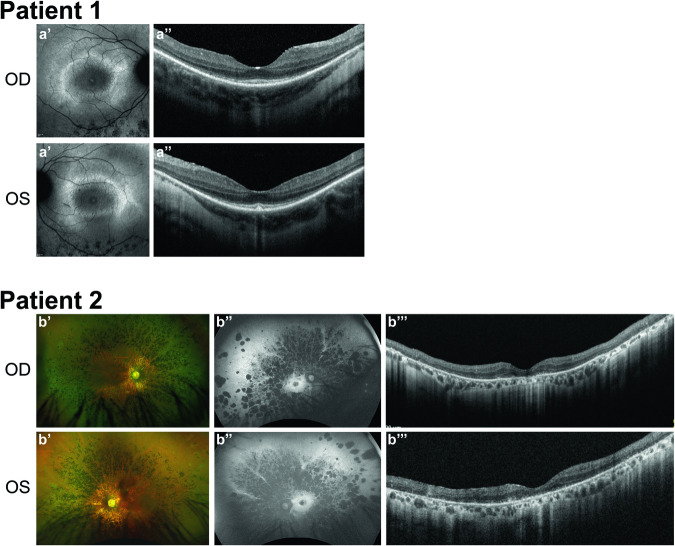
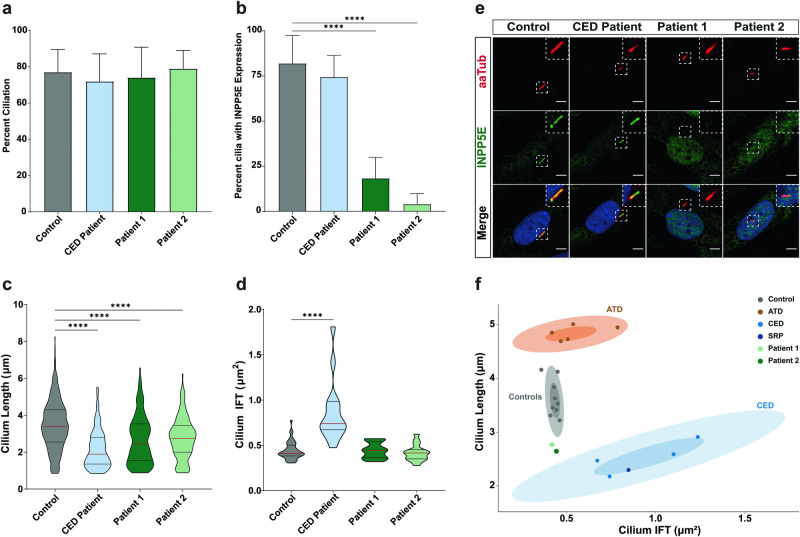
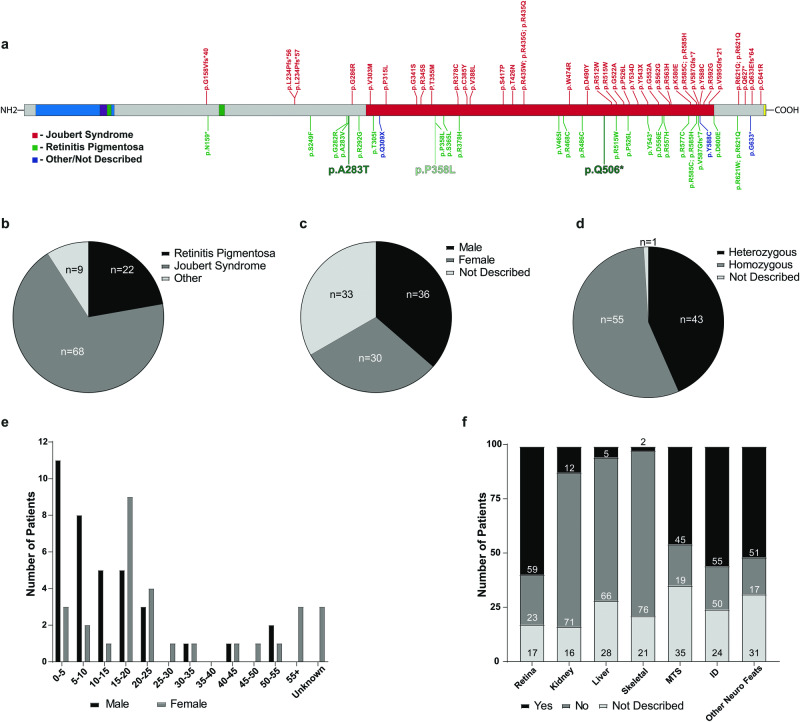
INPP5E encodes inositol polyphosphate-5-phosphatase E, an enzyme involved in regulating the phosphatidylinositol (PIP) makeup of the primary cilium membrane. Pathogenic variants in INPP5E hence cause a variety of ciliopathies: genetic disorders caused by dysfunctional cilia. While the majority of these disorders are syndromic, such as the neuronal ciliopathy Joubert syndrome, in some cases patients will present with an isolated phenotype-most commonly non-syndromic retinitis pigmentosa (RP). Here, we report two novel variants in INPP5E identified in two patients with non-syndromic RP: patient 1 with compound heterozygous variants (c.1516C > T, p.(Q506*), and c.847G > A, p.(A283T)) and patient 2 with a homozygous variant (c.1073C > T, p.(P358L)). To determine whether these variants were causative for the phenotype in the patients, automated ciliary phenotyping of patient-derived dermal fibroblasts was performed for percent ciliation, cilium length, retrograde IFT trafficking, and INPP5E localization. In both patients, a decrease in ciliary length and loss of INPP5E localization in the primary cilia were seen. With these molecular findings, we can confirm functionally that the novel variants in INPP5E are causative for the RP phenotypes seen in both patients. Additionally, this study demonstrates the usefulness of utilizing ciliary phenotyping as an assistant in ciliopathy diagnosis and phenotyping.
© 2024. The Author(s).
Conflict of interest statement
Figures
References
-
- Daiger SP, Sullivan LS, Bowne SJ. RetNet. Retinal Information Network. Available from: https://sph.uth.edu/retnet/.
-
- Georgiou M, Fujinami K, Michaelides M. Inherited retinal diseases: therapeutics, clinical trials and end points—a review. Clin Exp Ophthalmol. 2021;49:270–88. - PubMed
-
- Veltel S, Gasper R, Eisenacher E, Wittinghofer A. The retinitis pigmentosa 2 gene product is a GTPase-activating protein for Arf-like 3. Nat Struct Mol Biol. 2008;15:373–80. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
