

Single-cell RNA sequencing reveals the evolution of the immune landscape during perihematomal edema progression after intracerebral hemorrhage
- PMID: 38807233
- PMCID: PMC11131315
- DOI: 10.1186/s12974-024-03113-8
Single-cell RNA sequencing reveals the evolution of the immune landscape during perihematomal edema progression after intracerebral hemorrhage
Abstract
Background: Perihematomal edema (PHE) after post-intracerebral hemorrhage (ICH) has complex pathophysiological mechanisms that are poorly understood. The complicated immune response in the post-ICH brain constitutes a crucial component of PHE pathophysiology. In this study, we aimed to characterize the transcriptional profiles of immune cell populations in human PHE tissue and explore the microscopic differences between different types of immune cells.
Methods: 9 patients with basal ganglia intracerebral hemorrhage (hematoma volume 50-100 ml) were enrolled in this study. A multi-stage profile was developed, comprising Group1 (n = 3, 0-6 h post-ICH, G1), Group2 (n = 3, 6-24 h post-ICH, G2), and Group3 (n = 3, 24-48 h post-ICH, G3). A minimal quantity of edematous tissue surrounding the hematoma was preserved during hematoma evacuation. Single cell RNA sequencing (scRNA-seq) was used to map immune cell populations within comprehensively resected PHE samples collected from patients at different stages after ICH.
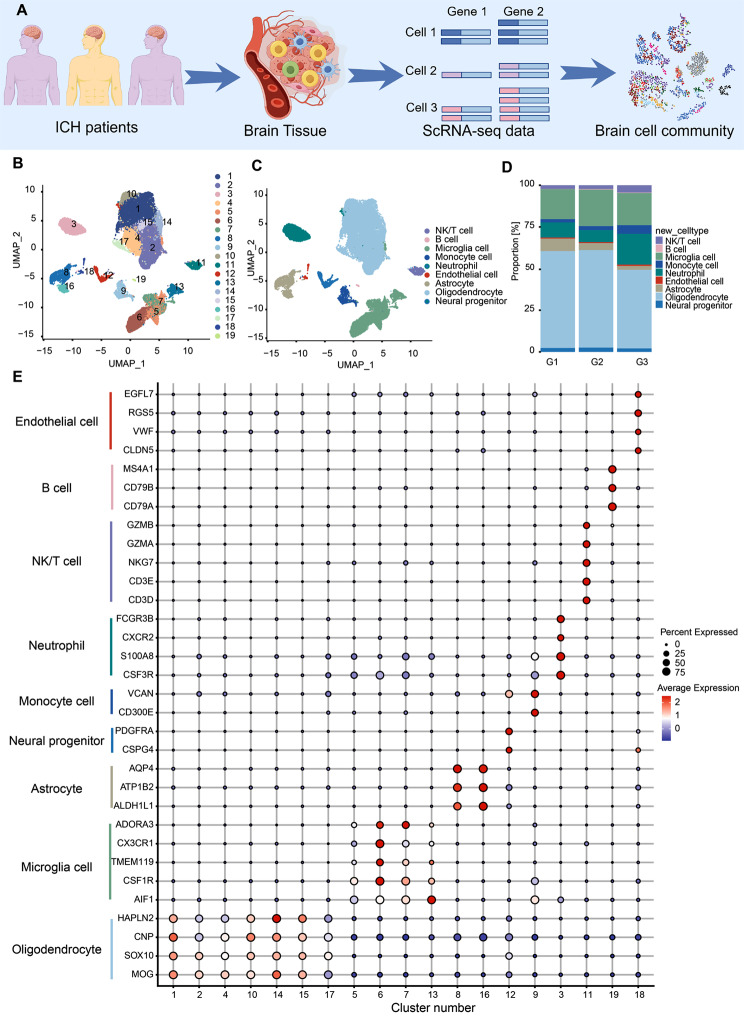
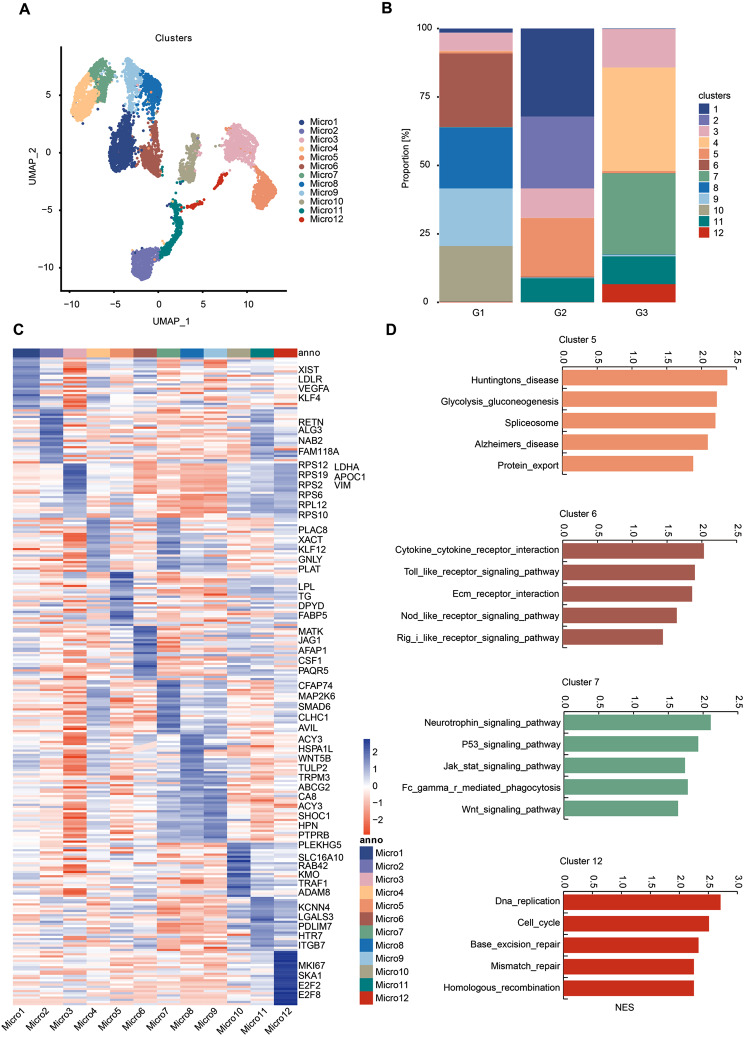
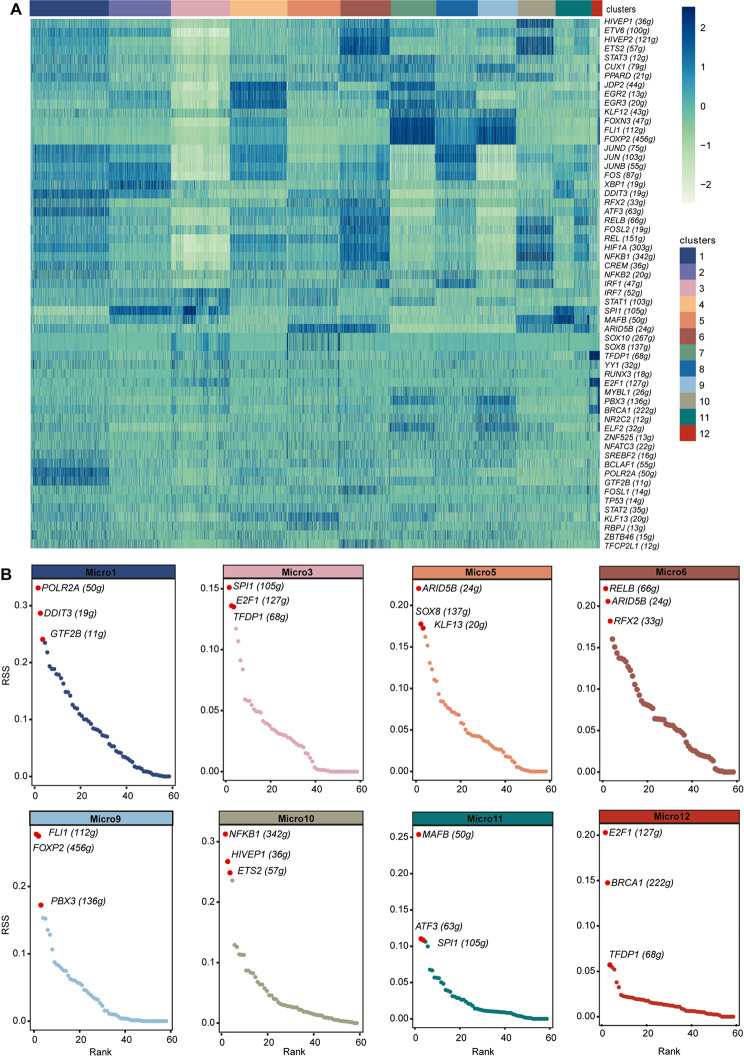
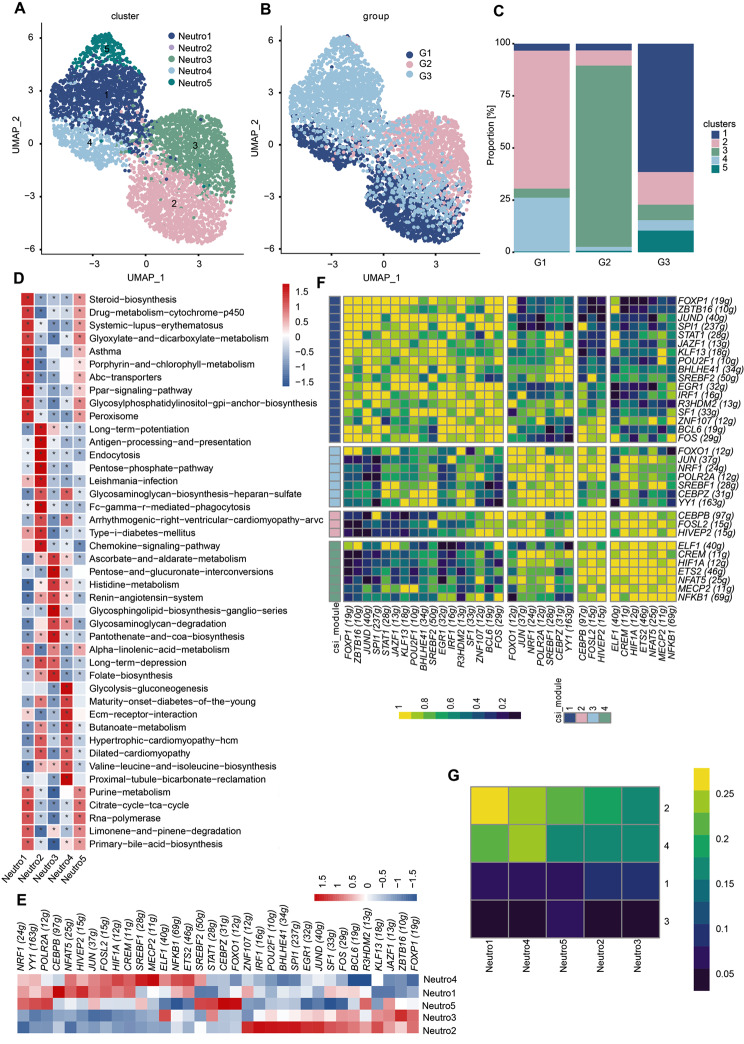
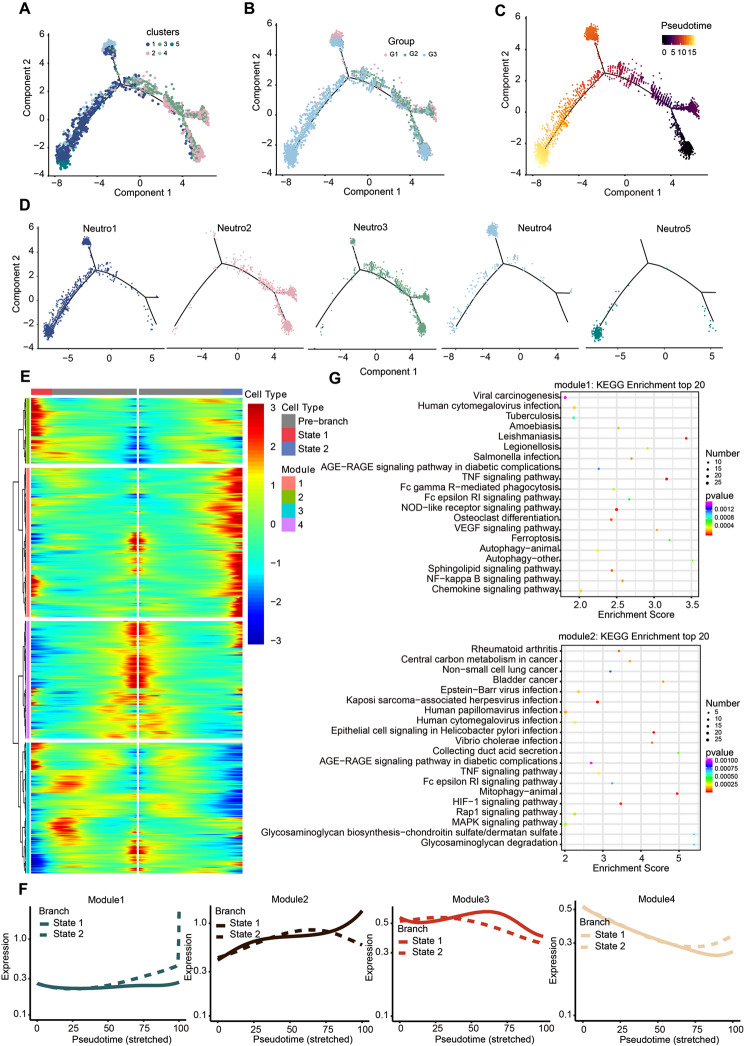
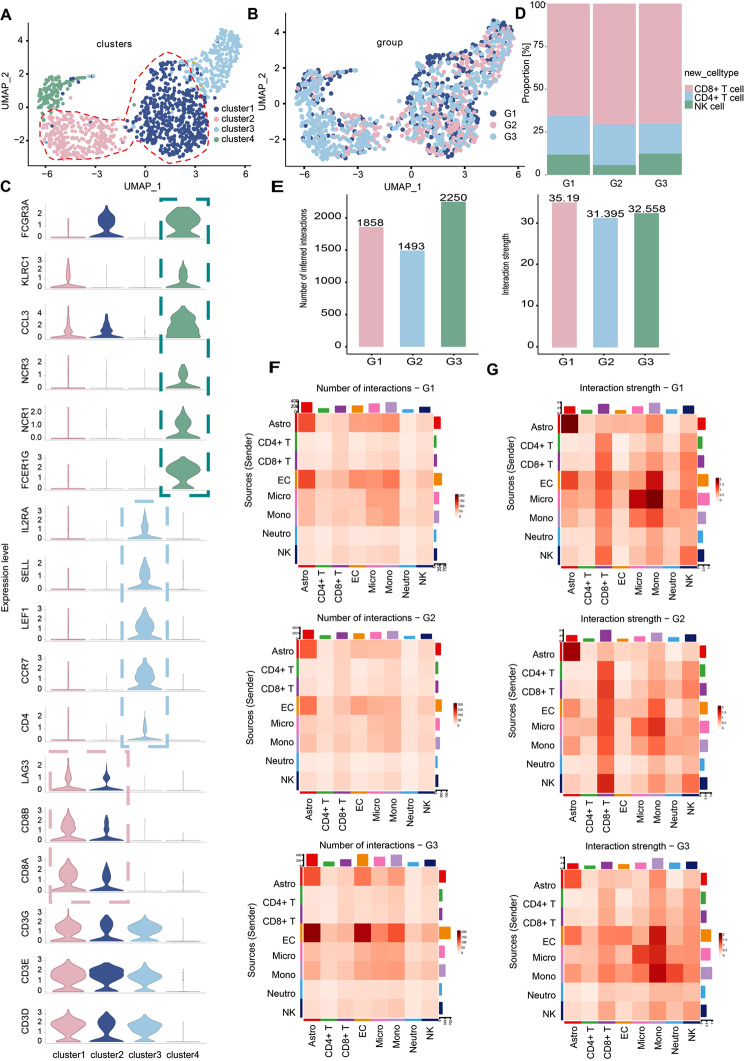
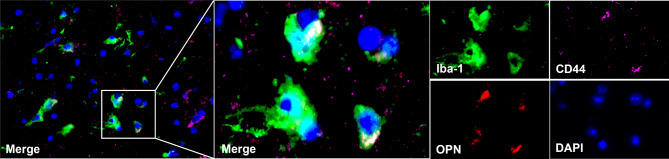
Results: We established, for the first time, a comprehensive landscape of diverse immune cell populations in human PHE tissue at a single-cell level. Our study identified 12 microglia subsets and 5 neutrophil subsets in human PHE tissue. What's more, we discovered that the secreted phosphoprotein-1 (SPP1) pathway served as the basis for self-communication between microglia subclusters during the progression of PHE. Additionally, we traced the trajectory branches of different neutrophil subtypes. Finally, we also demonstrated that microglia-produced osteopontin (OPN) could regulate the immune environment in PHE tissue by interacting with CD44-positive cells.
Conclusions: As a result of our research, we have gained valuable insight into the immune-microenvironment within PHE tissue, which could potentially be used to develop novel treatment modalities for ICH.
Keywords: Immune cell; Intracerebral hemorrhage; Osteopontin; Perihematomal edema; Single cell RNA sequencing.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures



References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
