

Pairtools: From sequencing data to chromosome contacts
- PMID: 38809952
- PMCID: PMC11164360
- DOI: 10.1371/journal.pcbi.1012164
Pairtools: From sequencing data to chromosome contacts
Abstract
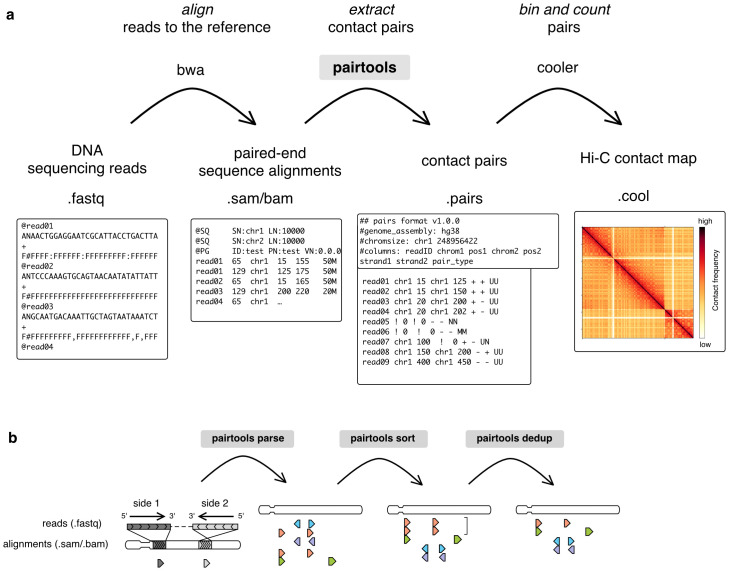
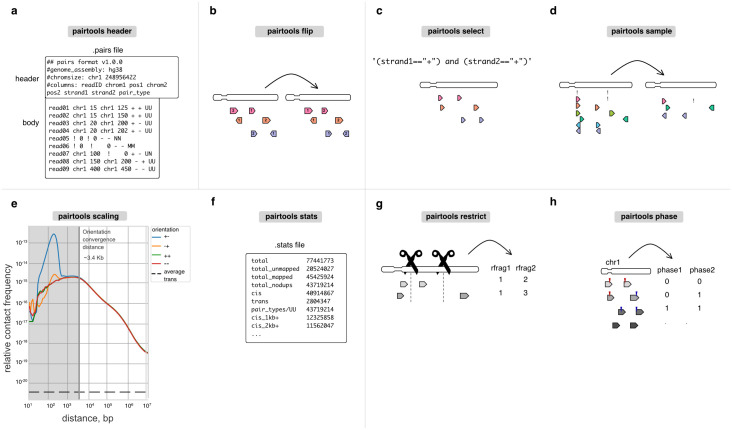
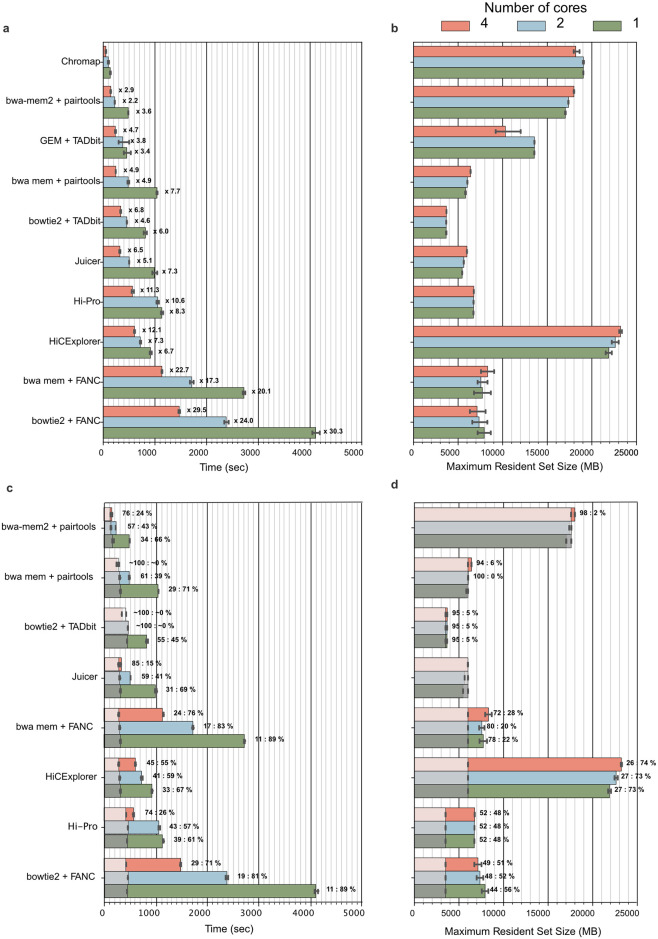
The field of 3D genome organization produces large amounts of sequencing data from Hi-C and a rapidly-expanding set of other chromosome conformation protocols (3C+). Massive and heterogeneous 3C+ data require high-performance and flexible processing of sequenced reads into contact pairs. To meet these challenges, we present pairtools-a flexible suite of tools for contact extraction from sequencing data. Pairtools provides modular command-line interface (CLI) tools that can be flexibly chained into data processing pipelines. The core operations provided by pairtools are parsing of.sam alignments into Hi-C pairs, sorting and removal of PCR duplicates. In addition, pairtools provides auxiliary tools for building feature-rich 3C+ pipelines, including contact pair manipulation, filtration, and quality control. Benchmarking pairtools against popular 3C+ data pipelines shows advantages of pairtools for high-performance and flexible 3C+ analysis. Finally, pairtools provides protocol-specific tools for restriction-based protocols, haplotype-resolved contacts, and single-cell Hi-C. The combination of CLI tools and tight integration with Python data analysis libraries makes pairtools a versatile foundation for a broad range of 3C+ pipelines.
Copyright: © 2024 Open2C et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Update of
-
Pairtools: from sequencing data to chromosome contacts.bioRxiv [Preprint]. 2023 Feb 15:2023.02.13.528389. doi: 10.1101/2023.02.13.528389. bioRxiv. 2023. Update in: PLoS Comput Biol. 2024 May 29;20(5):e1012164. doi: 10.1371/journal.pcbi.1012164. PMID: 36824968 Free PMC article. Updated. Preprint.
References
-
- Cohen NM, Olivares-Chauvet P, Lubling Y, Baran Y, Lifshitz A, Hoichman M, et al. SHAMAN: bin-free randomization, normalization and screening of Hi-C matrices. bioRxiv. 2017. p. 187203. doi: 10.1101/187203 - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
