

Structure-based discovery of CFTR potentiators and inhibitors
- PMID: 38810646
- PMCID: PMC11262615
- DOI: 10.1016/j.cell.2024.04.046
Structure-based discovery of CFTR potentiators and inhibitors
Abstract
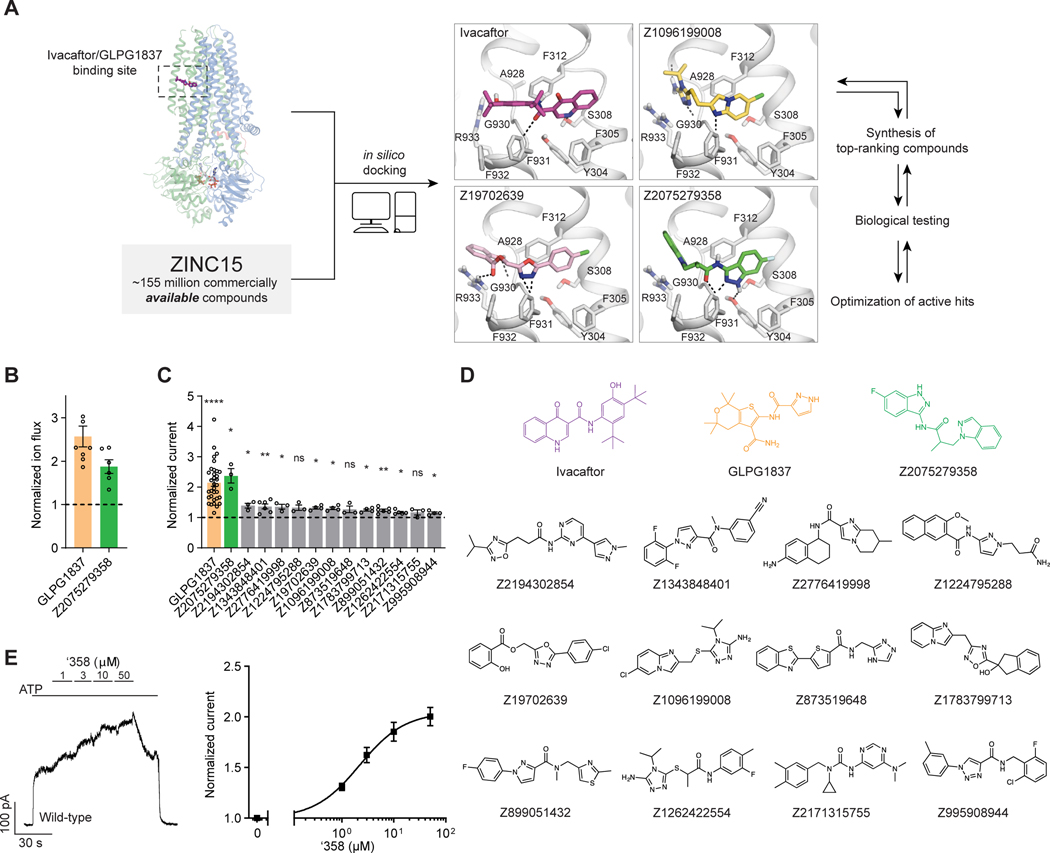
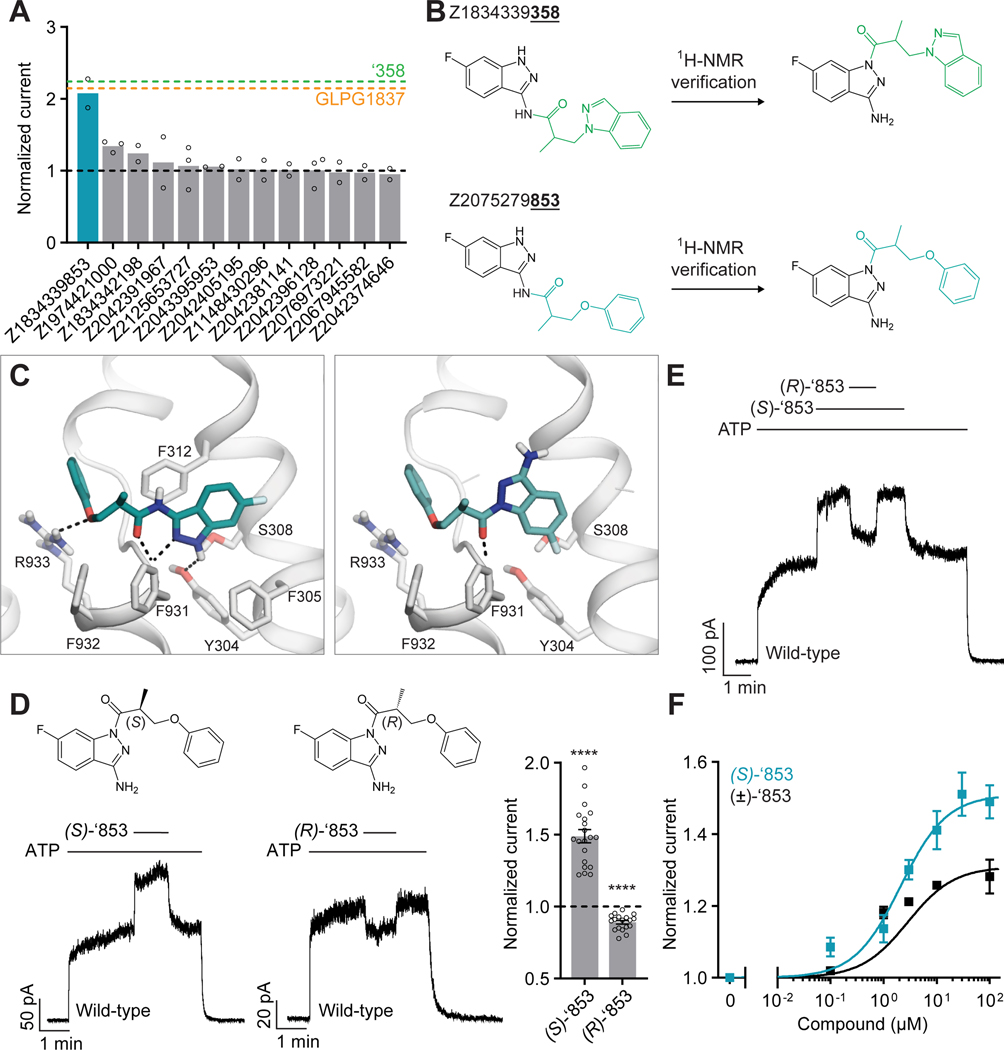
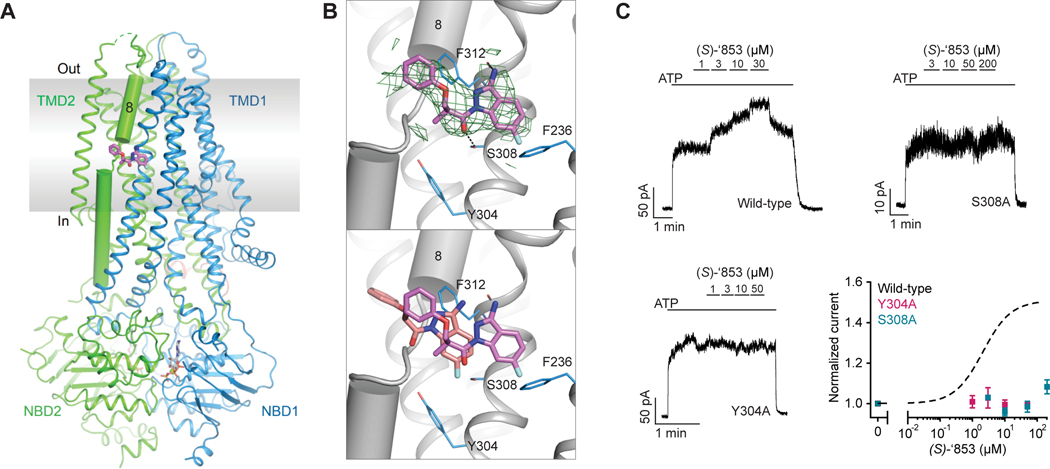
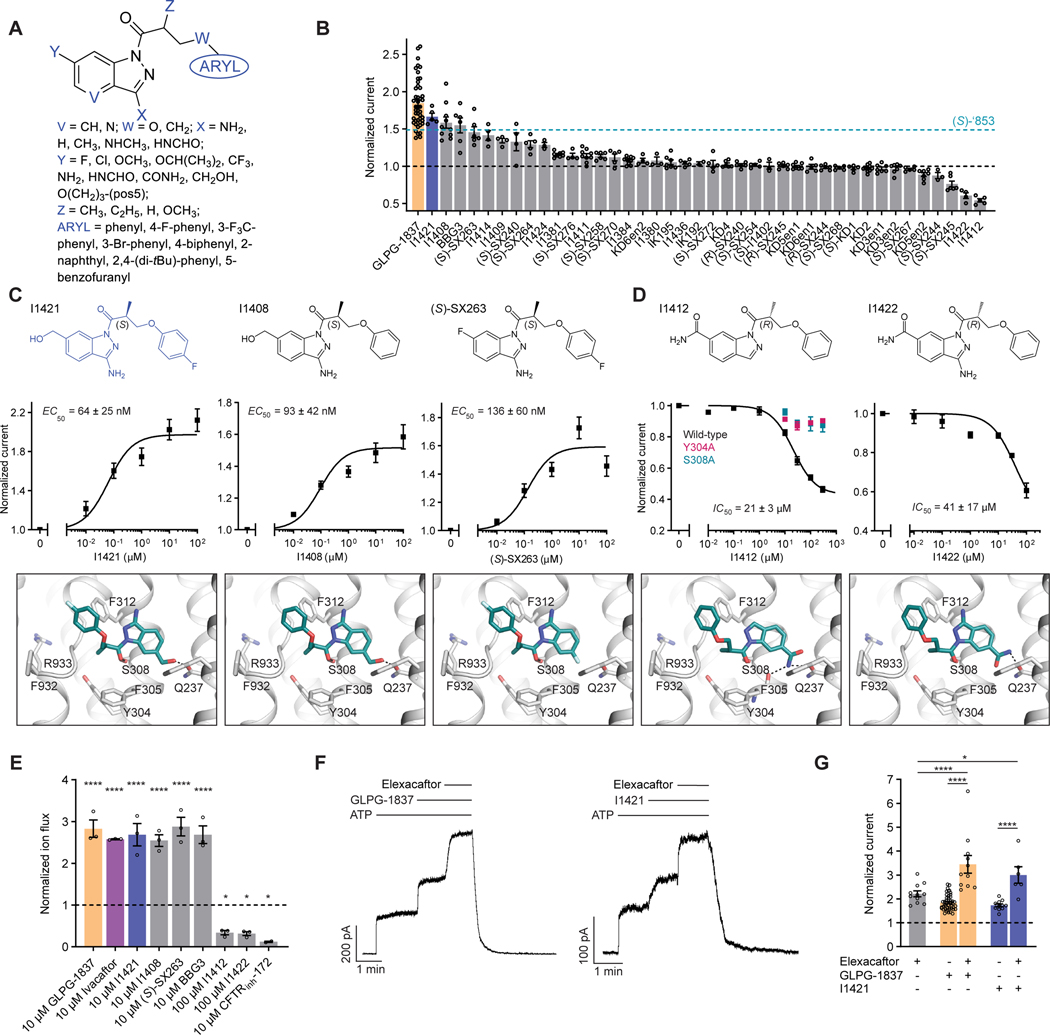
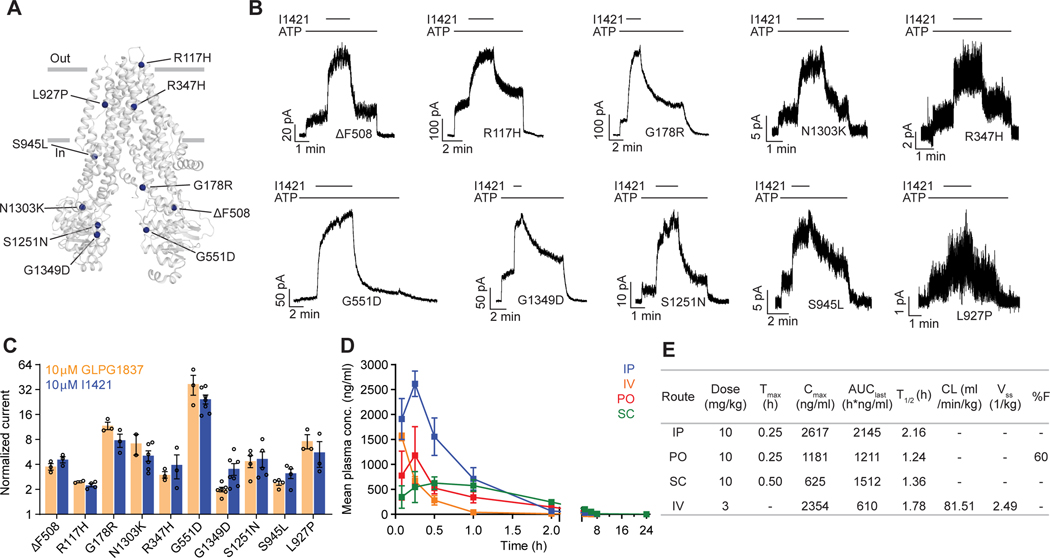
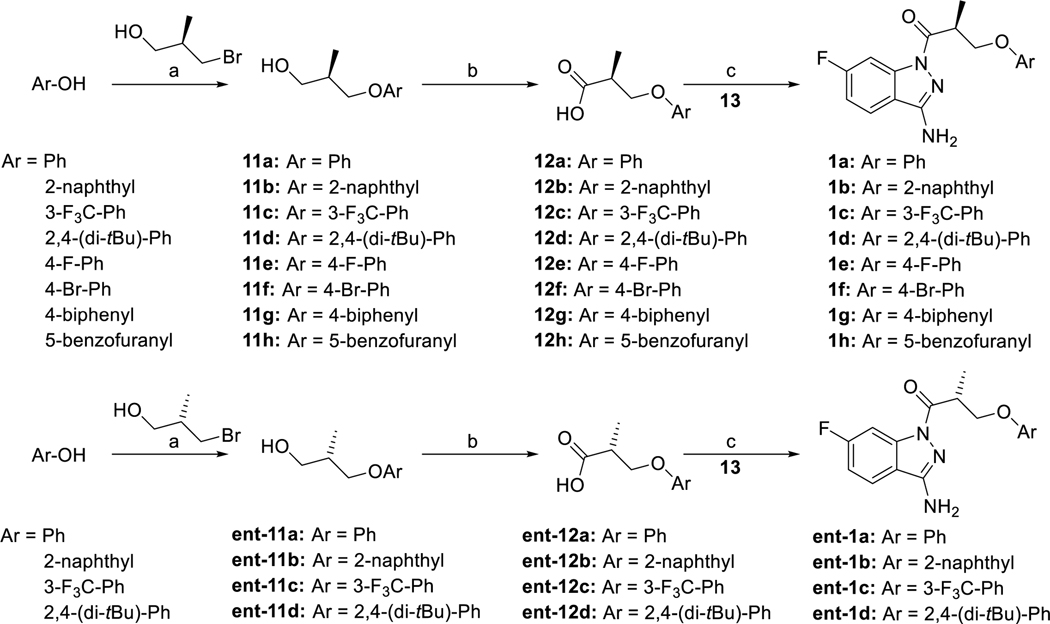
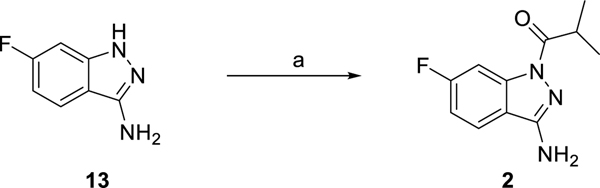
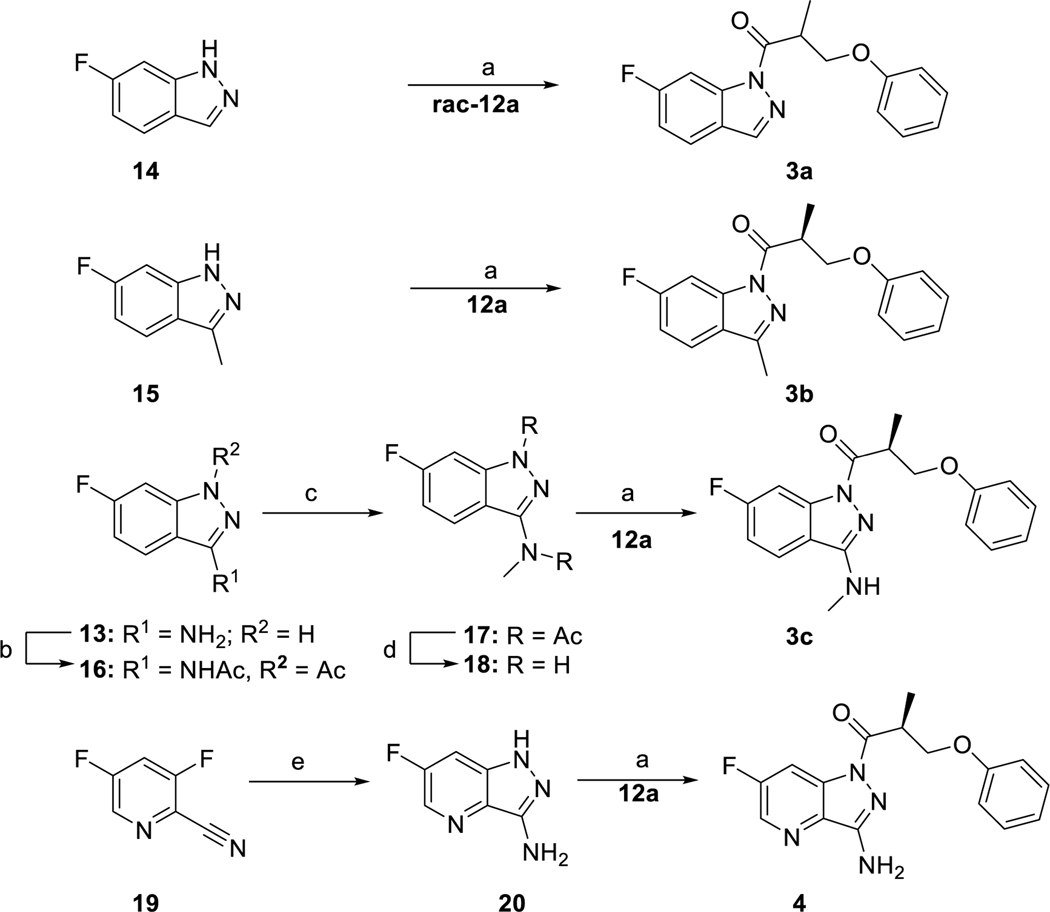
The cystic fibrosis transmembrane conductance regulator (CFTR) is a crucial ion channel whose loss of function leads to cystic fibrosis, whereas its hyperactivation leads to secretory diarrhea. Small molecules that improve CFTR folding (correctors) or function (potentiators) are clinically available. However, the only potentiator, ivacaftor, has suboptimal pharmacokinetics and inhibitors have yet to be clinically developed. Here, we combine molecular docking, electrophysiology, cryo-EM, and medicinal chemistry to identify CFTR modulators. We docked ∼155 million molecules into the potentiator site on CFTR, synthesized 53 test ligands, and used structure-based optimization to identify candidate modulators. This approach uncovered mid-nanomolar potentiators, as well as inhibitors, that bind to the same allosteric site. These molecules represent potential leads for the development of more effective drugs for cystic fibrosis and secretory diarrhea, demonstrating the feasibility of large-scale docking for ion channel drug discovery.
Keywords: ABC transporter; anion channel; inhibitors; large-scale docking; ligand discovery; potentiators.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests B.K.S. and P.G. are founders of Epiodyne. B.K.S. is a co-founder of BlueDolphin and Deep Apple Therapeutics, as is J.J.I., and serves on the SRB of Genentech and the SABs of Vilya Therapeutics and Umbra Therapeutics and consults for Great Point Ventures and Levator Therapeutics. A patent on the discovery of positive and negative allosteric regulators for CFTR has been filed. The authors declare no other competing interests.
Figures
Update of
-
Structure-based discovery of CFTR potentiators and inhibitors.bioRxiv [Preprint]. 2024 Mar 11:2023.09.09.557002. doi: 10.1101/2023.09.09.557002. bioRxiv. 2024. Update in: Cell. 2024 Jul 11;187(14):3712-3725.e34. doi: 10.1016/j.cell.2024.04.046. PMID: 37745391 Free PMC article. Updated. Preprint.
Comment in
-
Discovering CFTR modulators.Nat Rev Drug Discov. 2024 Aug;23(8):581. doi: 10.1038/d41573-024-00112-5. Nat Rev Drug Discov. 2024. PMID: 38937614 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
