

Biological basis of critical illness subclasses: from the bedside to the bench and back again
- PMID: 38812006
- PMCID: PMC11137966
- DOI: 10.1186/s13054-024-04959-3
Biological basis of critical illness subclasses: from the bedside to the bench and back again
Abstract
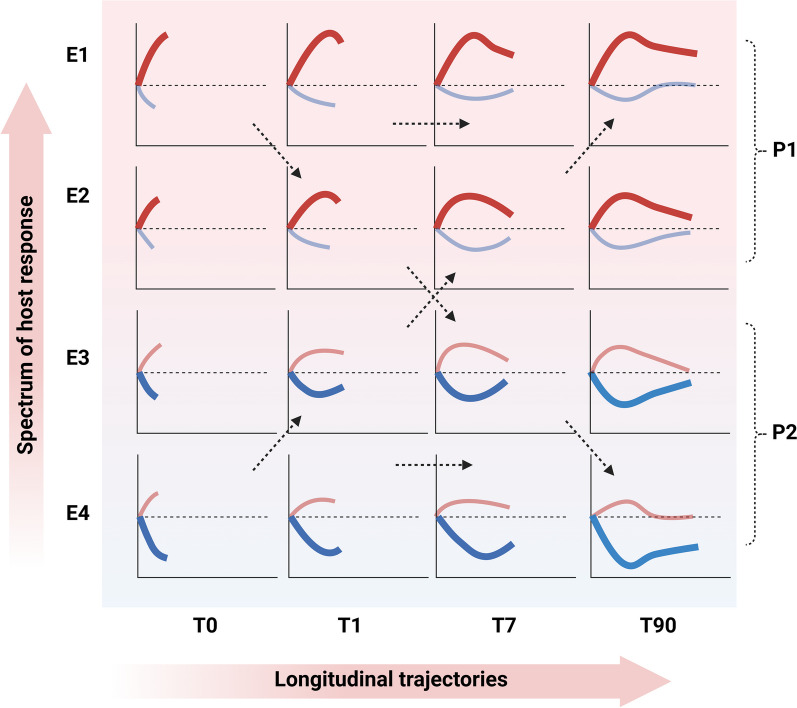
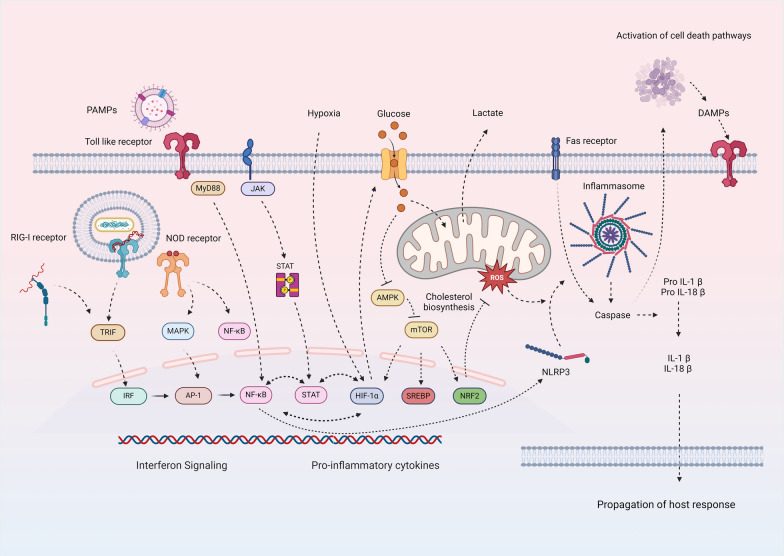
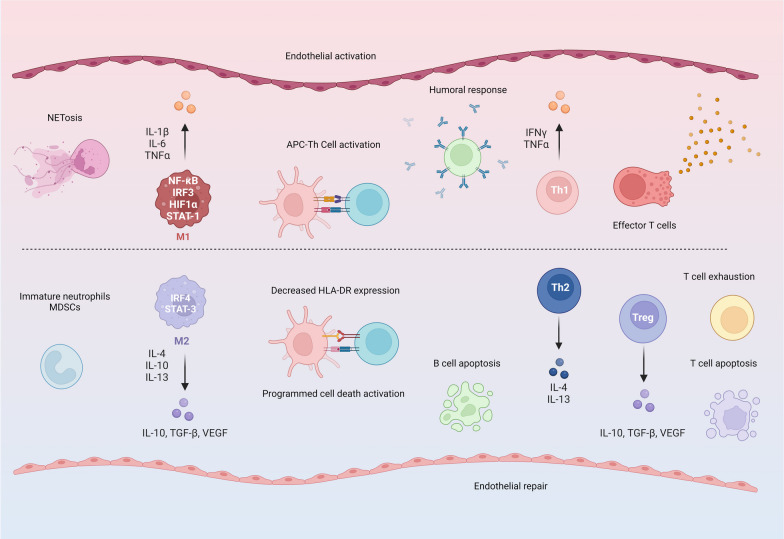
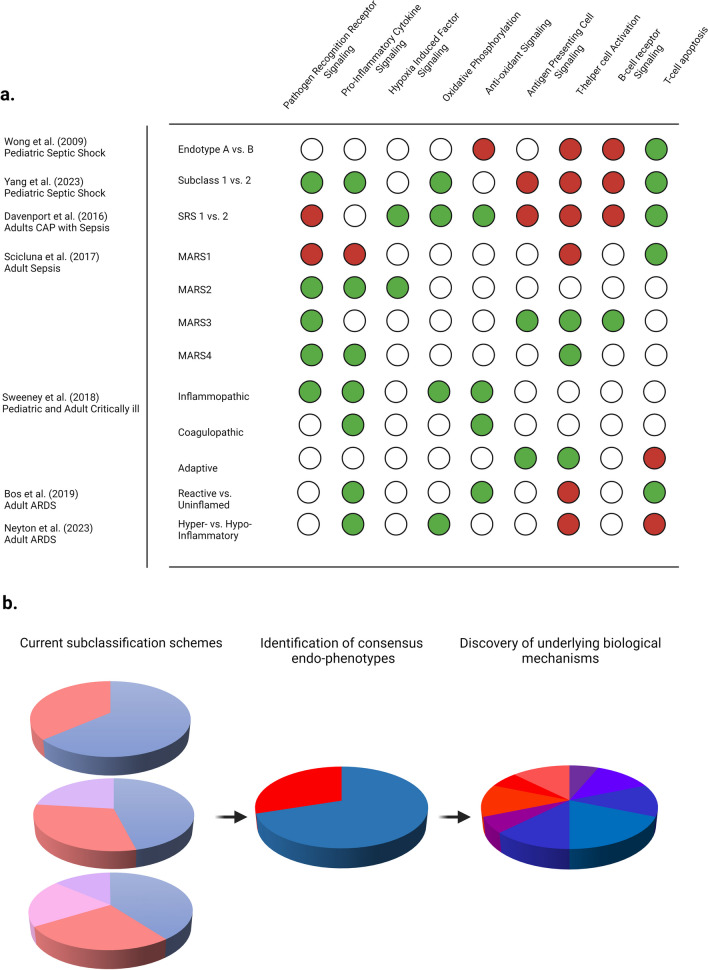
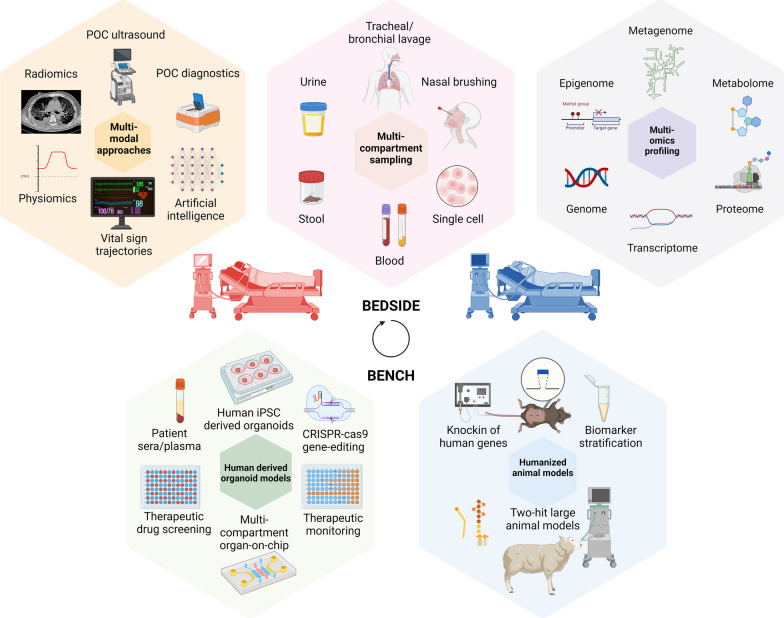
Critical illness syndromes including sepsis, acute respiratory distress syndrome, and acute kidney injury (AKI) are associated with high in-hospital mortality and long-term adverse health outcomes among survivors. Despite advancements in care, clinical and biological heterogeneity among patients continues to hamper identification of efficacious therapies. Precision medicine offers hope by identifying patient subclasses based on clinical, laboratory, biomarker and 'omic' data and potentially facilitating better alignment of interventions. Within the previous two decades, numerous studies have made strides in identifying gene-expression based endotypes and clinico-biomarker based phenotypes among critically ill patients associated with differential outcomes and responses to treatment. In this state-of-the-art review, we summarize the biological similarities and differences across the various subclassification schemes among critically ill patients. In addition, we highlight current translational gaps, the need for advanced scientific tools, human-relevant disease models, to gain a comprehensive understanding of the molecular mechanisms underlying critical illness subclasses.
Keywords: Critical illness subclass; Endotype; Phenotype; Precision medicine.
© 2024. The Author(s).
Conflict of interest statement
Cincinnati Children’s Hospital Medical Center (CCHMC) and the estate of the late Dr. Hector R. Wong hold patents for the pediatric sepsis biomarker risk model (PERSEVERE) for risk-stratification of pediatric sepsis patients and gene-expression based adaptive endotypes. M.R.A and CCHMC hold provisional patents for provisional patents (1) for a unified biomarker model – PERSEVERENCE that incorporates PERSEVERE and endothelial dysfunction markers to predict risk of multiple organ dysfunctions in sepsis, (2) a provisional patent for a PERSEVERENCE SA-AKI model that incorporates endothelial biomarkers predictive of persistent sepsis associated acute kidney injury for enrichment in clinical trials, and (3) a provisional patent for gene-expression-based multiple organ dysfunction syndrome (MODS) subclass identification, reflective of the host innate immune response.
Figures
Similar articles
-
Subphenotypes in critical care: translation into clinical practice.Lancet Respir Med. 2020 Jun;8(6):631-643. doi: 10.1016/S2213-2600(20)30124-7. Lancet Respir Med. 2020. PMID: 32526190 Review.
-
Prognostic and predictive enrichment in sepsis.Nat Rev Nephrol. 2020 Jan;16(1):20-31. doi: 10.1038/s41581-019-0199-3. Epub 2019 Sep 11. Nat Rev Nephrol. 2020. PMID: 31511662 Free PMC article. Review.
-
Lipid Mediators in Critically Ill Patients: A Step Towards Precision Medicine.Front Immunol. 2020 Nov 25;11:599853. doi: 10.3389/fimmu.2020.599853. eCollection 2020. Front Immunol. 2020. PMID: 33324417 Free PMC article. Review.
-
Laying the Groundwork for Physiology-Guided Precision Medicine in the Critically Ill.NEJM Evid. 2023 May;2(5):EVIDe2300051. doi: 10.1056/EVIDe2300051. Epub 2023 Apr 25. NEJM Evid. 2023. PMID: 38320026
-
Precision management of acute kidney injury in the intensive care unit: current state of the art.Intensive Care Med. 2023 Sep;49(9):1049-1061. doi: 10.1007/s00134-023-07171-z. Epub 2023 Aug 8. Intensive Care Med. 2023. PMID: 37552332 Review.
Cited by
-
Review of research progress in sepsis-associated acute kidney injury.Front Mol Biosci. 2025 Jul 11;12:1603392. doi: 10.3389/fmolb.2025.1603392. eCollection 2025. Front Mol Biosci. 2025. PMID: 40718792 Free PMC article. Review.
-
International multi-cohort analysis identifies novel framework for quantifying immune dysregulation in critical illness: results of the SUBSPACE consortium.bioRxiv [Preprint]. 2024 Nov 15:2024.11.12.623298. doi: 10.1101/2024.11.12.623298. bioRxiv. 2024. PMID: 39605502 Free PMC article. Preprint.
-
Heart-lung crosstalk in acute respiratory distress syndrome.Front Physiol. 2024 Oct 18;15:1478514. doi: 10.3389/fphys.2024.1478514. eCollection 2024. Front Physiol. 2024. PMID: 39493867 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
