

The short-chain fatty acid propionate prevents ox-LDL-induced coronary microvascular dysfunction by alleviating endoplasmic reticulum stress in HCMECs
- PMID: 38814895
- PMCID: PMC11139260
- DOI: 10.1371/journal.pone.0304551
The short-chain fatty acid propionate prevents ox-LDL-induced coronary microvascular dysfunction by alleviating endoplasmic reticulum stress in HCMECs
Abstract
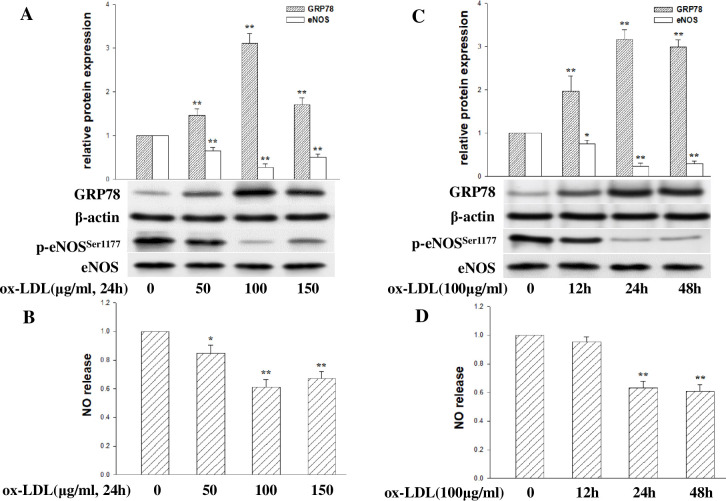
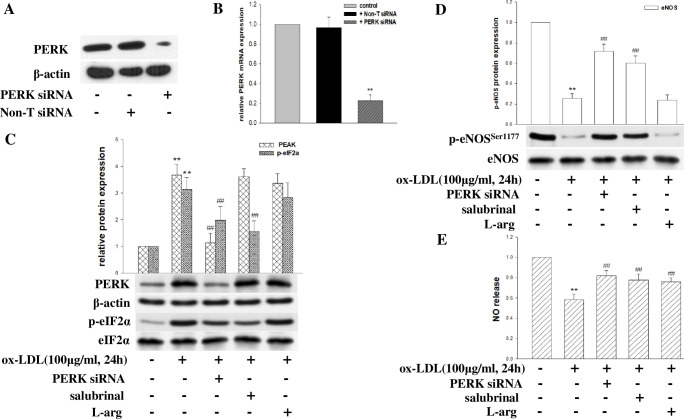
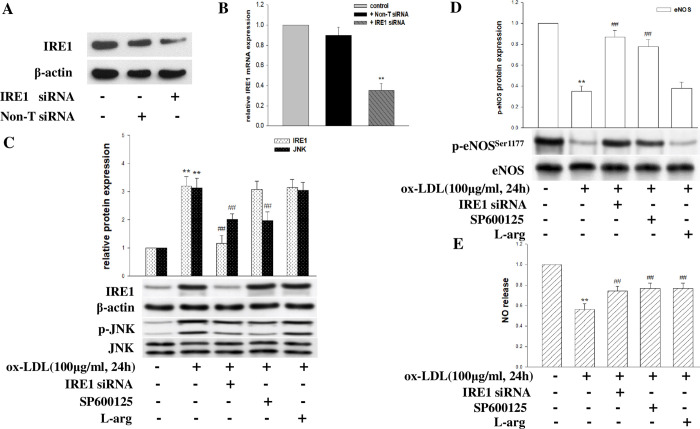
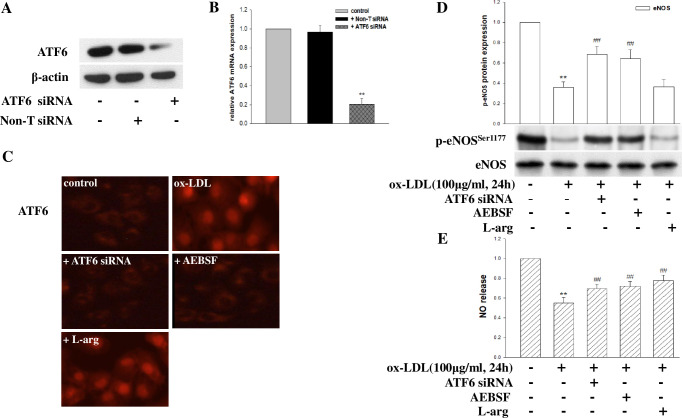
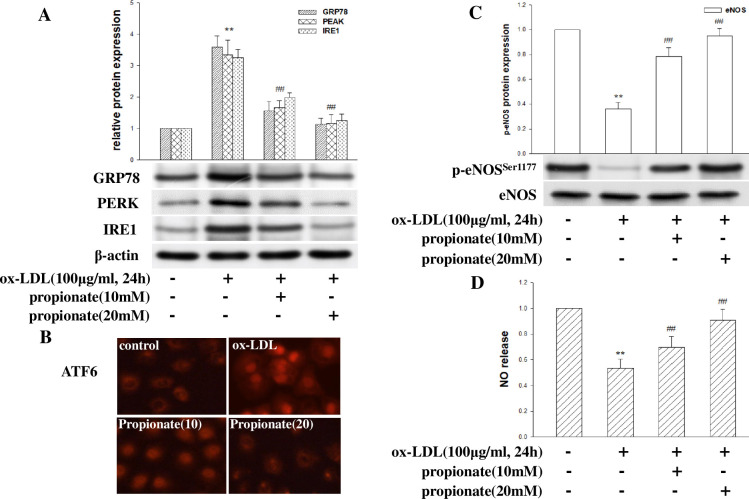
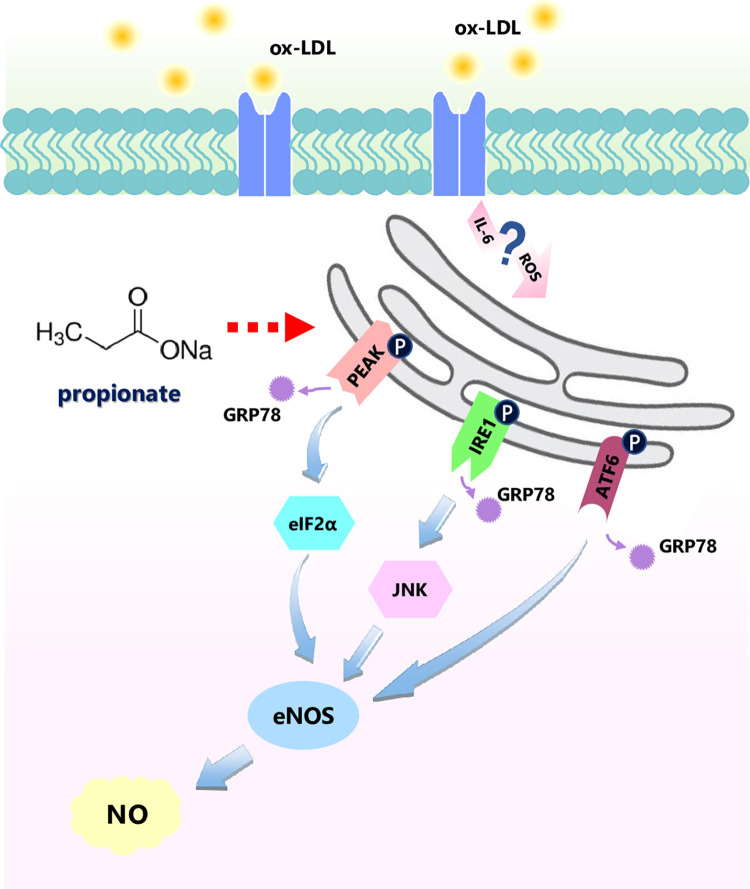
Coronary microvascular dysfunction (CMD) is a critical pathogenesis of cardiovascular diseases. Lower endothelial nitric oxide synthase (eNOS) phosphorylation leads to reduced endothelium-derived relaxing factor nitric oxide (NO) generation, causing and accelerating CMD. Endoplasmic reticulum stress (ER stress) has been shown to reduce NO production in umbilical vein endothelial cells. Oxidized low-density lipoprotein (ox-LDL) damages endothelial cell function. However, the relationship between ox-LDL and coronary microcirculation has yet to be assessed. Short-chain fatty acid (SCFA), a fermentation product of the gut microbiome, could improve endothelial-dependent vasodilation in human adipose arterioles, but the effect of SCFA on coronary microcirculation is unclear. In this study, we found ox-LDL stimulated expression of ER chaperone GRP78. Further, we activated downstream PERK/eIF2a, IRE1/JNK, and ATF6 signaling pathways, decreasing eNOS phosphorylation and NO production in human cardiac microvascular endothelial. Furthermore, SCFA-propionate can inhibit ox-LDL-induced eNOS phosphorylation reduction and raise NO production; the mechanism is related to the inhibition of ER stress and downstream signaling pathways PERK/eIF2a, IRE1/JNK, and ATF6. In summary, we demonstrate that ox-LDL induced CMD by activating ER stress, propionate can effectively counteract the adverse effects of ox-LDL and protect coronary microcirculation function via inhibiting ER stress.
Copyright: © 2024 Hong et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Endoplasmic reticulum stress promotes macrophage-derived foam cell formation by up-regulating cluster of differentiation 36 (CD36) expression.J Biol Chem. 2014 Feb 14;289(7):4032-42. doi: 10.1074/jbc.M113.524512. Epub 2013 Dec 23. J Biol Chem. 2014. PMID: 24366867 Free PMC article.
-
ox-LDL downregulates eNOS activity via LOX-1-mediated endoplasmic reticulum stress.Int J Mol Med. 2013 Dec;32(6):1442-50. doi: 10.3892/ijmm.2013.1513. Epub 2013 Sep 30. Int J Mol Med. 2013. PMID: 24085225
-
Effect of anti-hyperglycemic drugs on endoplasmic reticulum (ER) stress in human coronary artery endothelial cells.Eur J Pharmacol. 2021 Sep 15;907:174249. doi: 10.1016/j.ejphar.2021.174249. Epub 2021 Jun 9. Eur J Pharmacol. 2021. PMID: 34116042
-
Molecular signal networks and regulating mechanisms of the unfolded protein response.J Zhejiang Univ Sci B. 2017 Jan.;18(1):1-14. doi: 10.1631/jzus.B1600043. J Zhejiang Univ Sci B. 2017. PMID: 28070992 Free PMC article. Review.
-
Navigating the Landscape of Coronary Microvascular Research: Trends, Triumphs, and Challenges Ahead.Rev Cardiovasc Med. 2024 Aug 16;25(8):288. doi: 10.31083/j.rcm2508288. eCollection 2024 Aug. Rev Cardiovasc Med. 2024. PMID: 39228508 Free PMC article. Review.
Cited by
-
Berberine and its derivatives: mechanisms of action in myocardial vascular endothelial injury - a review.Front Pharmacol. 2025 Mar 4;16:1543697. doi: 10.3389/fphar.2025.1543697. eCollection 2025. Front Pharmacol. 2025. PMID: 40103596 Free PMC article. Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
