

MSL2 variants lead to a neurodevelopmental syndrome with lack of coordination, epilepsy, specific dysmorphisms, and a distinct episignature
- PMID: 38815585
- PMCID: PMC11267526
- DOI: 10.1016/j.ajhg.2024.05.001
MSL2 variants lead to a neurodevelopmental syndrome with lack of coordination, epilepsy, specific dysmorphisms, and a distinct episignature
Abstract
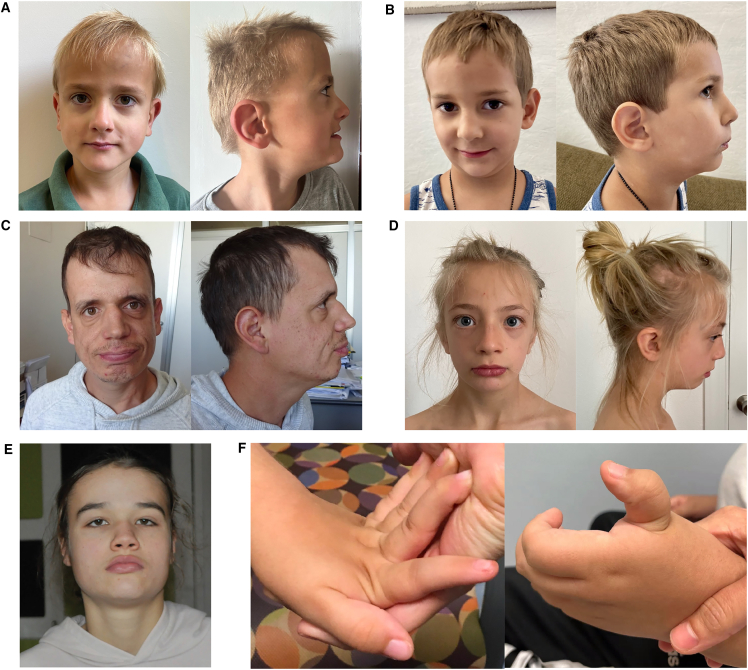
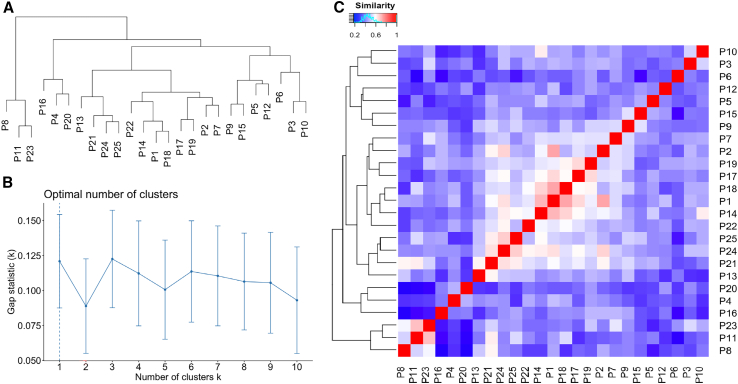
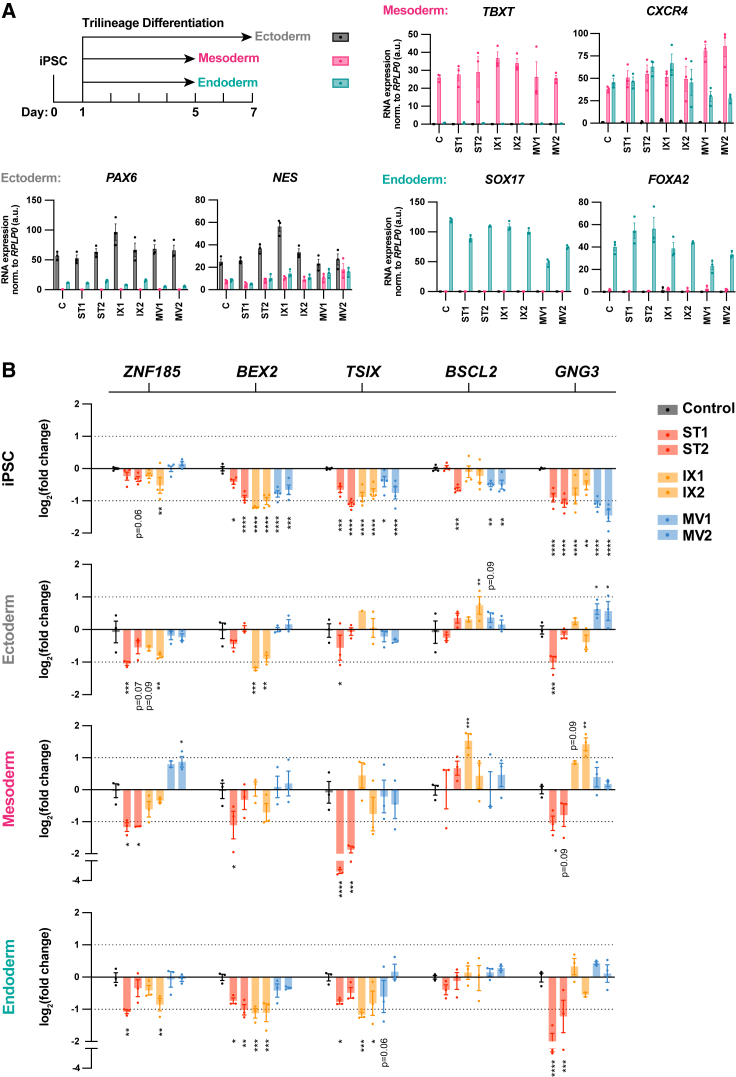
Epigenetic dysregulation has emerged as an important etiological mechanism of neurodevelopmental disorders (NDDs). Pathogenic variation in epigenetic regulators can impair deposition of histone post-translational modifications leading to aberrant spatiotemporal gene expression during neurodevelopment. The male-specific lethal (MSL) complex is a prominent multi-subunit epigenetic regulator of gene expression and is responsible for histone 4 lysine 16 acetylation (H4K16ac). Using exome sequencing, here we identify a cohort of 25 individuals with heterozygous de novo variants in MSL complex member MSL2. MSL2 variants were associated with NDD phenotypes including global developmental delay, intellectual disability, hypotonia, and motor issues such as coordination problems, feeding difficulties, and gait disturbance. Dysmorphisms and behavioral and/or psychiatric conditions, including autism spectrum disorder, and to a lesser extent, seizures, connective tissue disease signs, sleep disturbance, vision problems, and other organ anomalies, were observed in affected individuals. As a molecular biomarker, a sensitive and specific DNA methylation episignature has been established. Induced pluripotent stem cells (iPSCs) derived from three members of our cohort exhibited reduced MSL2 levels. Remarkably, while NDD-associated variants in two other members of the MSL complex (MOF and MSL3) result in reduced H4K16ac, global H4K16ac levels are unchanged in iPSCs with MSL2 variants. Regardless, MSL2 variants altered the expression of MSL2 targets in iPSCs and upon their differentiation to early germ layers. Our study defines an MSL2-related disorder as an NDD with distinguishable clinical features, a specific blood DNA episignature, and a distinct, MSL2-specific molecular etiology compared to other MSL complex-related disorders.
Keywords: MSL2; autism; connective tissue; epigenetics; epilepsy; episignature; iPSC; male-specific lethal complex; neurodevelopmental syndrome.
Copyright © 2024. Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests B.S. is a shareholder in EpiSign Inc, involved in commercial uses of EpiSign(TM) technology D.A.C. and K.G.M. are employees of GeneDx, LLC.
Figures




References
-
- Radzisheuskaya A., Shliaha P.V., Grinev V.V., Shlyueva D., Damhofer H., Koche R., Gorshkov V., Kovalchuk S., Zhan Y., Rodriguez K.L., et al. Complex-dependent histone acetyltransferase activity of KAT8 determines its role in transcription and cellular homeostasis. Mol. Cell. 2021;81:1749–1765.e8. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
