

Primary cilia suppress the fibrotic activity of atrial fibroblasts from patients with atrial fibrillation in vitro
- PMID: 38816374
- PMCID: PMC11139955
- DOI: 10.1038/s41598-024-60298-x
Primary cilia suppress the fibrotic activity of atrial fibroblasts from patients with atrial fibrillation in vitro
Abstract
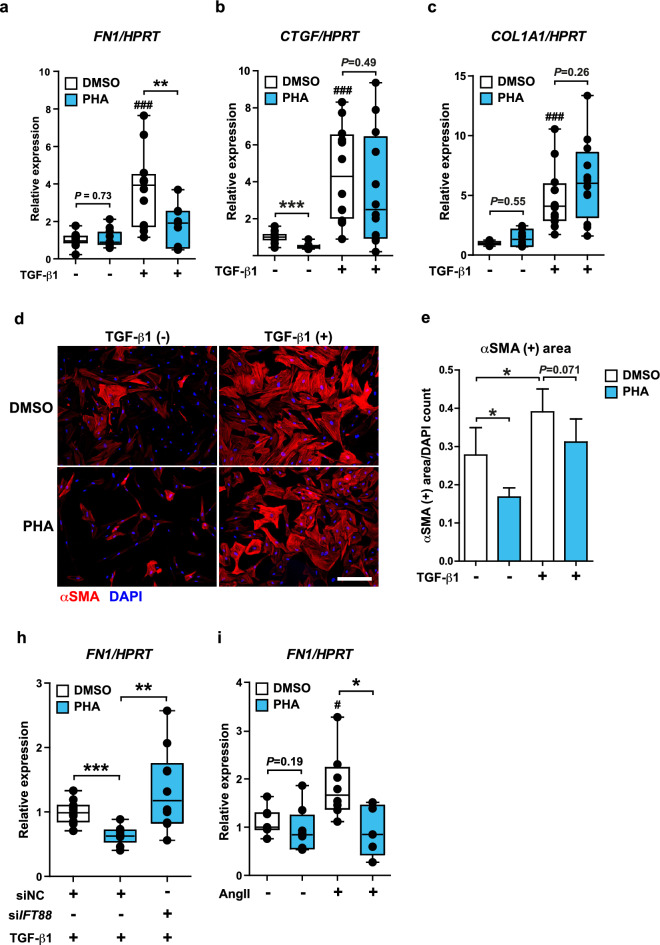
Atrial fibrosis serves as an arrhythmogenic substrate in atrial fibrillation (AF) and contributes to AF persistence. Treating atrial fibrosis is challenging because atrial fibroblast activity is multifactorial. We hypothesized that the primary cilium regulates the profibrotic response of AF atrial fibroblasts, and explored therapeutic potentials of targeting primary cilia to treat fibrosis in AF. We included 25 patients without AF (non-AF) and 26 persistent AF patients (AF). Immunohistochemistry using a subset of the patients (non-AF: n = 10, AF: n = 10) showed less ciliated fibroblasts in AF versus non-AF. Acetylated α-tubulin protein levels were decreased in AF, while the gene expressions of AURKA and NEDD9 were highly increased in AF patients' left atrium. Loss of primary cilia in human atrial fibroblasts through IFT88 knockdown enhanced expression of ECM genes, including FN1 and COL1A1. Remarkably, restoration or elongation of primary cilia by an AURKA selective inhibitor or lithium chloride, respectively, prevented the increased expression of ECM genes induced by different profibrotic cytokines in atrial fibroblasts of AF patients. Our data reveal a novel mechanism underlying fibrotic substrate formation via primary cilia loss in AF atrial fibroblasts and suggest a therapeutic potential for abrogating atrial fibrosis by restoring primary cilia.
© 2024. The Author(s).
Conflict of interest statement
JRdG received research grants through his institution from Abbott, Atricure, Boston Scientific, Bayer, Daiichi Sankyo, Johnson&Johnson, Medtronic Servier, and received speaker/consultancy fees from Atricure, Bayer, Daiichi Sankyo, Johnson&Johnson and Medtronic outside the submitted work. AHGD is a consultant for Atricure. The other authors report no disclosures.
Figures






References
-
- Thrall G, Lane D, Carroll D, Lip GYH. Quality of life in patients with atrial fibrillation: A systematic review. Am. J. Med. 2006;119(448):e1–448.e19. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
