

Systematic assessment of ISWI subunits shows that NURF creates local accessibility for CTCF
- PMID: 38816647
- PMCID: PMC11176080
- DOI: 10.1038/s41588-024-01767-x
Systematic assessment of ISWI subunits shows that NURF creates local accessibility for CTCF
Erratum in
-
Author Correction: Systematic assessment of ISWI subunits shows that NURF creates local accessibility for CTCF.Nat Genet. 2024 Nov;56(11):2574-2575. doi: 10.1038/s41588-024-01985-3. Nat Genet. 2024. PMID: 39487354 Free PMC article. No abstract available.
Abstract
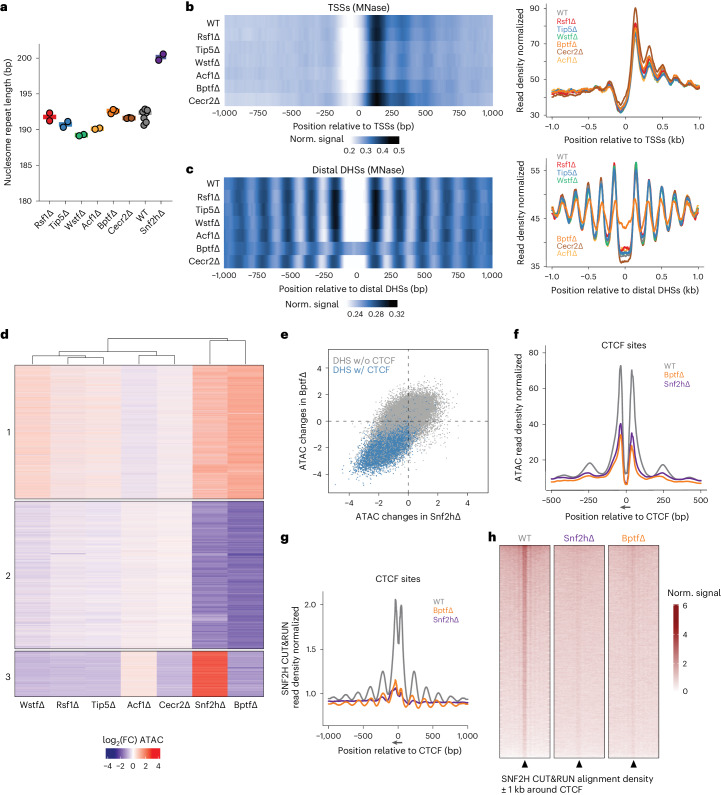
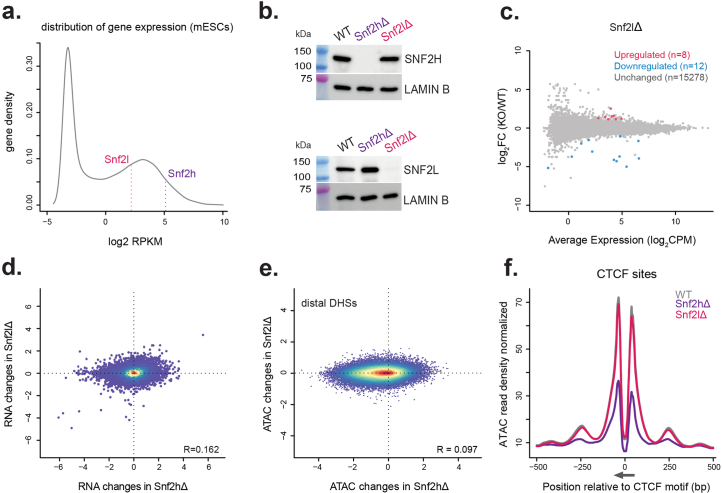
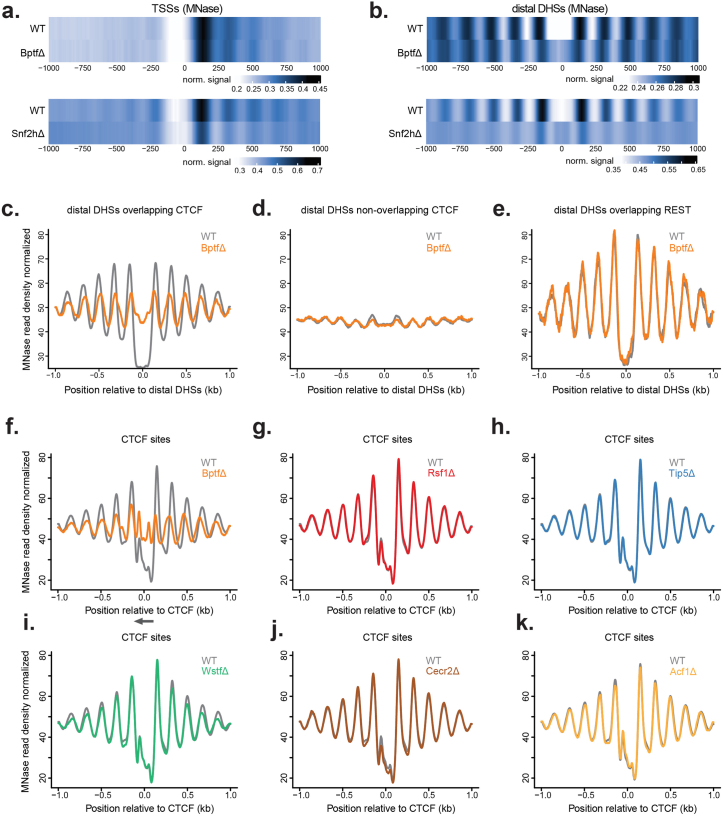
Catalytic activity of the imitation switch (ISWI) family of remodelers is critical for nucleosomal organization and DNA binding of certain transcription factors, including the insulator protein CTCF. Here we define the contribution of individual subcomplexes by deriving a panel of isogenic mouse stem cell lines, each lacking one of six ISWI accessory subunits. Individual deletions of subunits of either CERF, RSF, ACF, WICH or NoRC subcomplexes only moderately affect the chromatin landscape, while removal of the NURF-specific subunit BPTF leads to a strong reduction in chromatin accessibility and SNF2H ATPase localization around CTCF sites. This affects adjacent nucleosome occupancy and CTCF binding. At a group of sites with reduced chromatin accessibility, CTCF binding persists but cohesin occupancy is reduced, resulting in decreased insulation. These results suggest that CTCF binding can be separated from its function as an insulator in nuclear organization and identify a specific role for NURF in mediating SNF2H localization and chromatin opening at bound CTCF sites.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures












References
-
- Clapier, C. R. & Cairns, B. R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem.78, 273–304 (2009). - PubMed
-
- Bell, O., Tiwari, V. K., Thomä, N. H. & Schübeler, D. Determinants and dynamics of genome accessibility. Nat. Rev. Genet.12, 554–564 (2011). - PubMed
-
- Corona, D. F. & Tamkun, J. W. Multiple roles for ISWI in transcription, chromosome organization and DNA replication. Biochim. Biophys. Acta1677, 113–119 (2004). - PubMed
-
- Erdel, F. & Rippe, K. Chromatin remodelling in mammalian cells by ISWI-type complexes—where, when and why? FEBS J.278, 3608–3618 (2011). - PubMed
MeSH terms
Substances
Grants and funding
- 310030B_176394/Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (Swiss National Science Foundation)
- 310030_192642/Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (Swiss National Science Foundation)
- ReadMe-667951 and DNAaccess-884664/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- 759366, 'BioMeTre'/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- ALTF 611-2019/European Molecular Biology Organization (EMBO)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
