

Whole genome sequencing identifies elusive variants in genetically unsolved Italian inherited retinal disease patients
- PMID: 38816995
- PMCID: PMC11225895
- DOI: 10.1016/j.xhgg.2024.100314
Whole genome sequencing identifies elusive variants in genetically unsolved Italian inherited retinal disease patients
Abstract
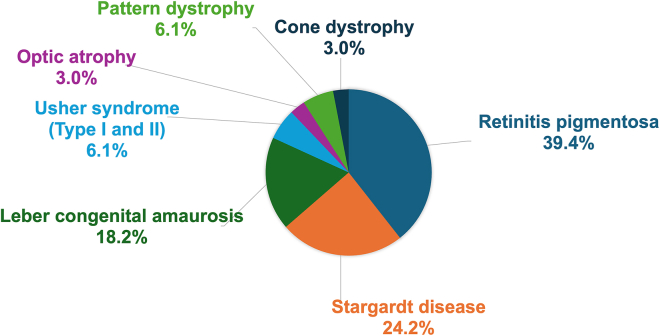
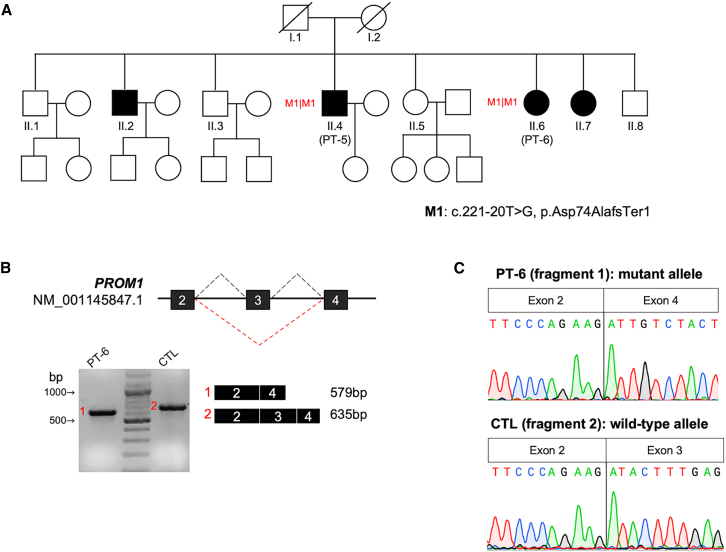
Inherited retinal diseases (IRDs) are a group of rare monogenic diseases with high genetic heterogeneity (pathogenic variants identified in over 280 causative genes). The genetic diagnostic rate for IRDs is around 60%, mainly thanks to the routine application of next-generation sequencing (NGS) approaches such as extensive gene panels or whole exome analyses. Whole-genome sequencing (WGS) has been reported to improve this diagnostic rate by revealing elusive variants, such as structural variants (SVs) and deep intronic variants (DIVs). We performed WGS on 33 unsolved cases with suspected autosomal recessive IRD, aiming to identify causative genetic variants in non-coding regions or to detect SVs that were unexplored in the initial screening. Most of the selected cases (30 of 33, 90.9%) carried monoallelic pathogenic variants in genes associated with their clinical presentation, hence we first analyzed the non-coding regions of these candidate genes. Whenever additional pathogenic variants were not identified with this approach, we extended the search for SVs and DIVs to all IRD-associated genes. Overall, we identified the missing causative variants in 11 patients (11 of 33, 33.3%). These included three DIVs in ABCA4, CEP290 and RPGRIP1; one non-canonical splice site (NCSS) variant in PROM1 and three SVs (large deletions) in EYS, PCDH15 and USH2A. For the previously unreported DIV in CEP290 and for the NCCS variant in PROM1, we confirmed the effect on splicing by reverse transcription (RT)-PCR on patient-derived RNA. This study demonstrates the power and clinical utility of WGS as an all-in-one test to identify disease-causing variants missed by standard NGS diagnostic methodologies.
Keywords: DIVs; IRD; SVs; WGS; deep intronic variants; inherited retinal diseases; structural variants; unsolved monoallelic cases.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures


References
-
- Berger W., Kloeckener-Gruissem B., Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010;29:335–375. - PubMed
-
- Schneider N., Sundaresan Y., Gopalakrishnan P., Beryozkin A., Hanany M., Levanon E.Y., Banin E., Ben-Aroya S., Sharon D. Inherited retinal diseases: Linking genes, disease-causing variants, and relevant therapeutic modalities. Prog. Retin. Eye Res. 2022;89 - PubMed
-
- Rivolta C., Sharon D., DeAngelis M.M., Dryja T.P. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum. Mol. Genet. 2002;11:1219–1227. - PubMed
-
- Eisenberger T., Neuhaus C., Khan A.O., Decker C., Preising M.N., Friedburg C., Bieg A., Gliem M., Charbel Issa P., Holz F.G., et al. Increasing the Yield in Targeted Next-Generation Sequencing by Implicating CNV Analysis, Non-Coding Exons and the Overall Variant Load: The Example of Retinal Dystrophies. Li T., editor. PLoS One. 2013;8 - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
