

Pgam5 aggravates hyperglycemia-induced myocardial dysfunction through disrupting Phb2-dependent mitochondrial dynamics
- PMID: 38818468
- PMCID: PMC11134593
- DOI: 10.7150/ijms.92872
Pgam5 aggravates hyperglycemia-induced myocardial dysfunction through disrupting Phb2-dependent mitochondrial dynamics
Abstract
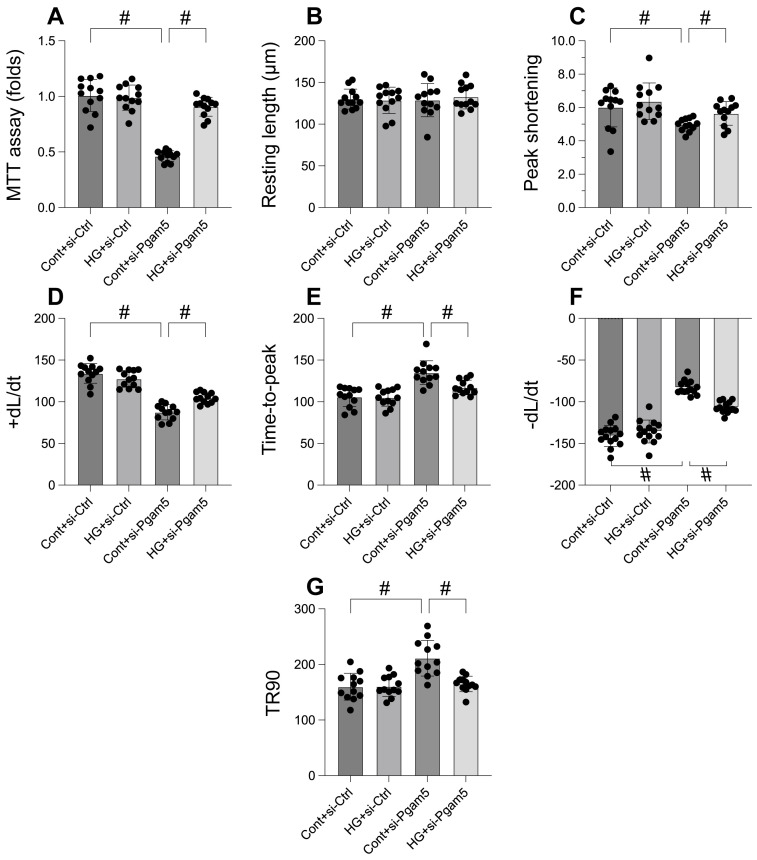
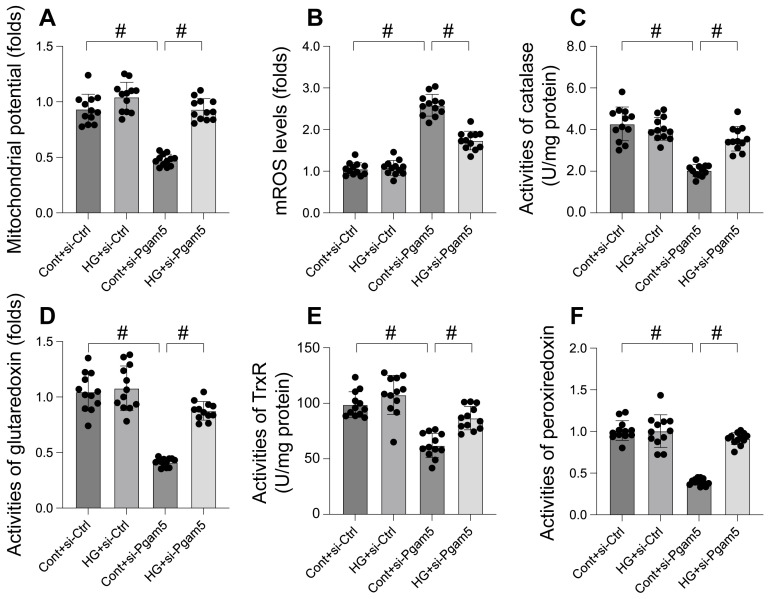
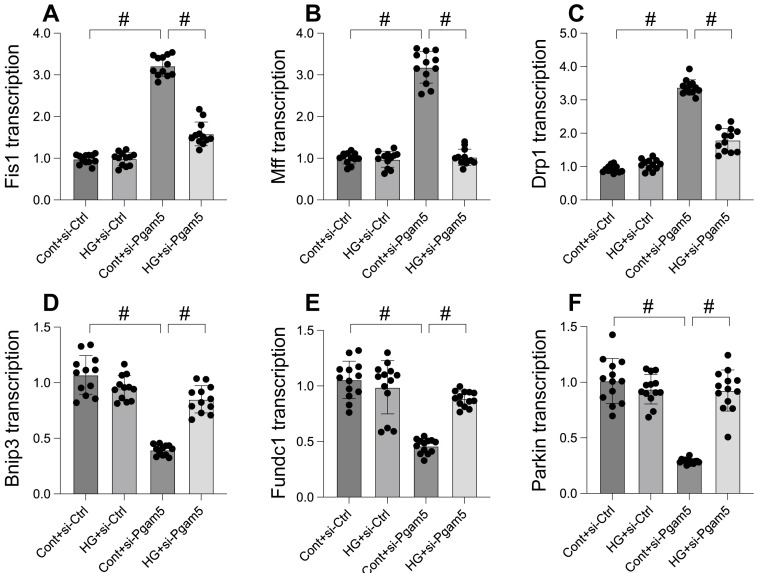
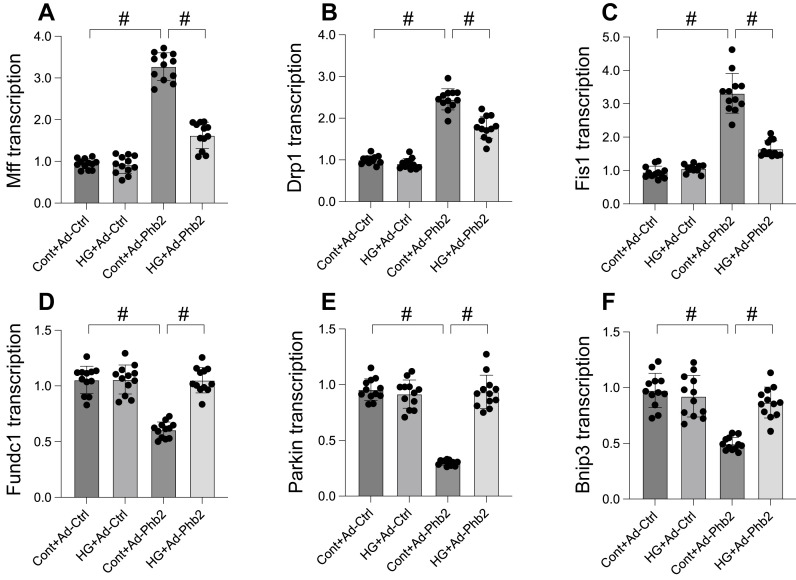
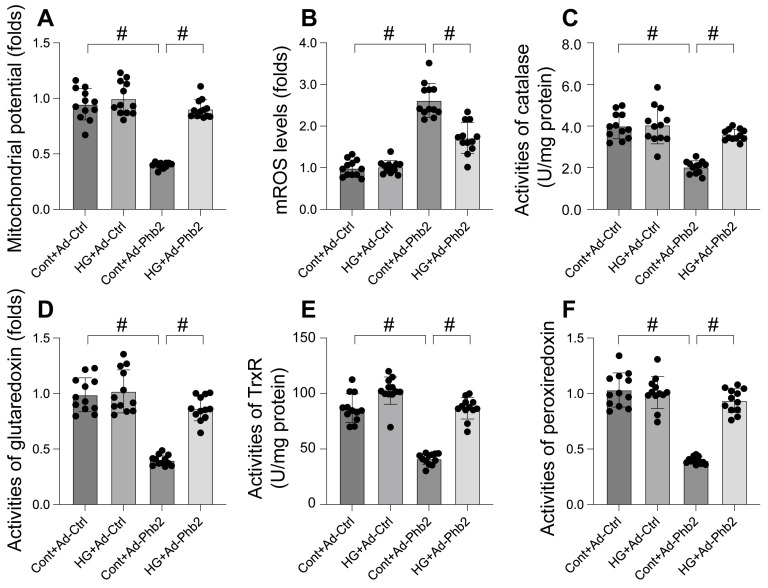
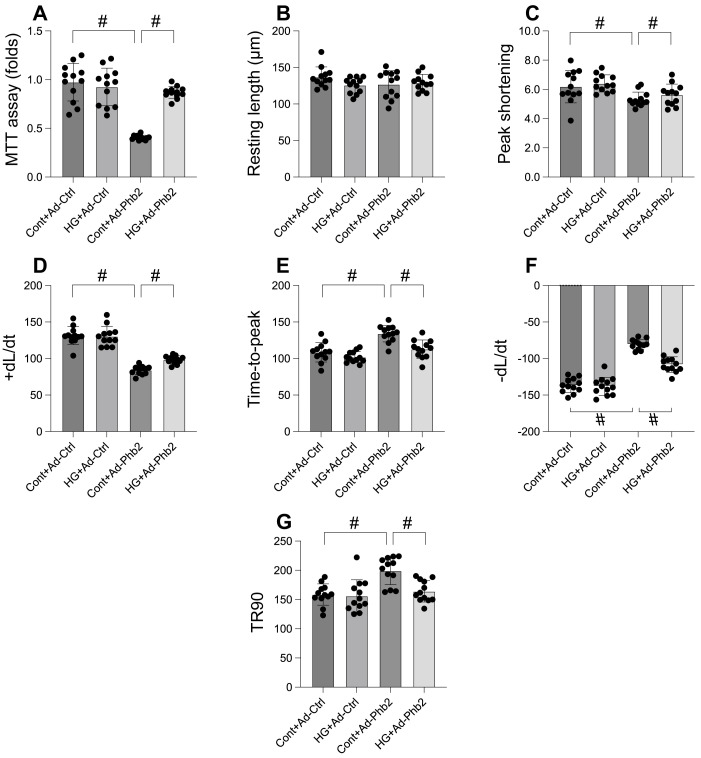
This study aims to elucidate the roles of Phosphoglycerate Mutase Family Member 5 (Pgam5) and Prohibitin 2 (Phb2) in the context of hyperglycemia-induced myocardial dysfunction, a critical aspect of diabetic cardiomyopathy. The research employed primary cardiomyocytes, which were then subjected to hyperglycemia treatment to mimic diabetic conditions. We used siRNA transfection to knock down Pgam5 and overexpressed Phb2 using adenovirus transfection to assess their individual and combined effects on cardiomyocyte health. Mitochondrial function was evaluated through measurements of mitochondrial membrane potential using the JC-1 probe, and levels of mitochondrial reactive oxygen species (ROS) were assessed. Additionally, the study involved qPCR analysis to quantify the transcriptional changes in genes related to mitochondrial fission and mitophagy. Our findings indicate that hyperglycemia significantly reduces cardiomyocyte viability and impairs mitochondrial function, as evidenced by decreased mitochondrial membrane potential and increased ROS levels. Pgam5 knockdown was observed to mitigate these adverse effects, preserving mitochondrial function and cardiomyocyte viability. On the molecular level, Pgam5 was found to regulate genes associated with mitochondrial fission (such as Drp1, Mff, and Fis1) and mitophagy (including Parkin, Bnip3, and Fundc1). Furthermore, overexpression of Phb2 countered the hyperglycemia-induced mitochondrial dysfunction and normalized the levels of key mitochondrial antioxidant enzymes. The combined data suggest a protective role for both Pgam5 knockdown and Phb2 overexpression against hyperglycemia-induced cellular and mitochondrial damage. The study elucidates the critical roles of Pgam5 and Phb2 in regulating mitochondrial dynamics in the setting of hyperglycemia-induced myocardial dysfunction. By modulating mitochondrial fission and mitophagy, Pgam5 and Phb2 emerge as key players in preserving mitochondrial integrity and cardiomyocyte health under diabetic conditions. These findings contribute significantly to our understanding of the molecular mechanisms underlying diabetic cardiomyopathy and suggest potential therapeutic targets for mitigating myocardial dysfunction in diabetes.
Keywords: Pgam5; Phb2; diabetic cardiomyopathy; mitochondrial fission; mitophagy.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
Similar articles
-
Phosphoglycerate mutase 5 exacerbates alcoholic cardiomyopathy in male mice by inducing prohibitin-2 dephosphorylation and impairing mitochondrial quality control.Clin Transl Med. 2024 Aug;14(8):e1806. doi: 10.1002/ctm2.1806. Clin Transl Med. 2024. PMID: 39143739 Free PMC article.
-
Pgam5-mediated PHB2 dephosphorylation contributes to endotoxemia-induced myocardial dysfunction by inhibiting mitophagy and the mitochondrial unfolded protein response.Int J Biol Sci. 2023 Aug 28;19(14):4657-4671. doi: 10.7150/ijbs.85767. eCollection 2023. Int J Biol Sci. 2023. PMID: 37781037 Free PMC article.
-
PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis.Autophagy. 2020 Mar;16(3):419-434. doi: 10.1080/15548627.2019.1628520. Epub 2019 Jun 16. Autophagy. 2020. PMID: 31177901 Free PMC article.
-
PGAM5: A crucial role in mitochondrial dynamics and programmed cell death.Eur J Cell Biol. 2021 Jan;100(1):151144. doi: 10.1016/j.ejcb.2020.151144. Epub 2020 Dec 7. Eur J Cell Biol. 2021. PMID: 33370650 Review.
-
Mitochondrial network remodeling of the diabetic heart: implications to ischemia related cardiac dysfunction.Cardiovasc Diabetol. 2024 Jul 18;23(1):261. doi: 10.1186/s12933-024-02357-1. Cardiovasc Diabetol. 2024. PMID: 39026280 Free PMC article. Review.
References
-
- Lorenzo-Almorós A, Cepeda-Rodrigo JM, Lorenzo Ó. Diabetic cardiomyopathy. Rev Clin Esp (Barc) 2022;222:100–11. - PubMed
-
- Lorenzo-Almorós A, Cepeda-Rodrigo JM, Lorenzo Ó. Diabetic cardiomyopathy. Rev Clin Esp. 2020. - PubMed
-
- Paolillo S, Marsico F, Prastaro M, Renga F, Esposito L, De Martino F. et al. Diabetic Cardiomyopathy: Definition, Diagnosis, and Therapeutic Implications. Heart Fail Clin. 2019;15:341–7. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
