

Mutations in the U4 snRNA gene RNU4-2 cause one of the most prevalent monogenic neurodevelopmental disorders
- PMID: 38821540
- PMCID: PMC11333284
- DOI: 10.1038/s41591-024-03085-5
Mutations in the U4 snRNA gene RNU4-2 cause one of the most prevalent monogenic neurodevelopmental disorders
Abstract
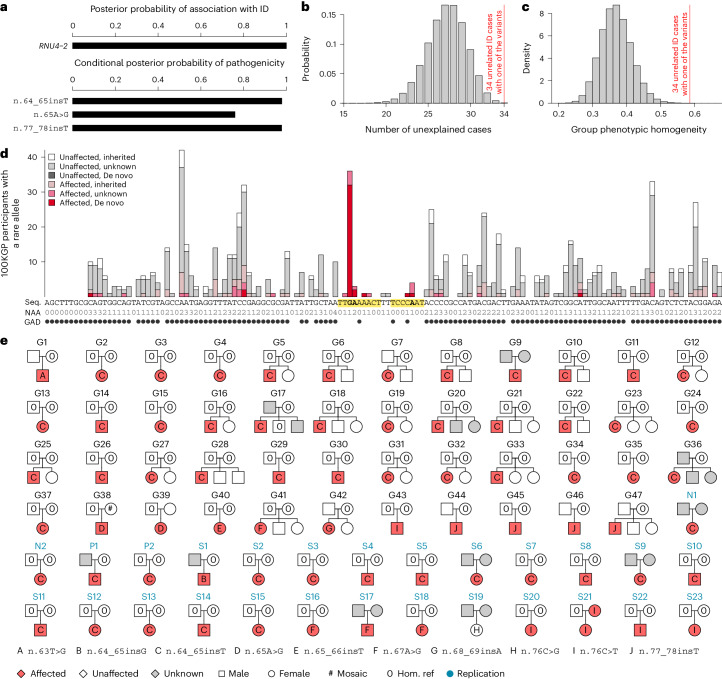
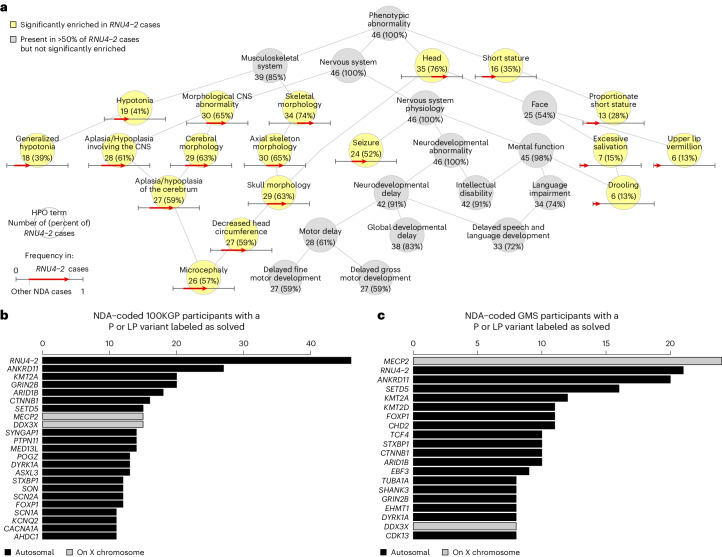
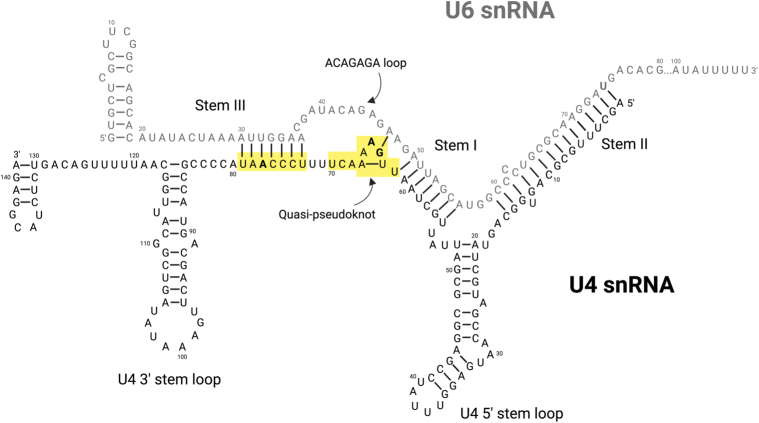
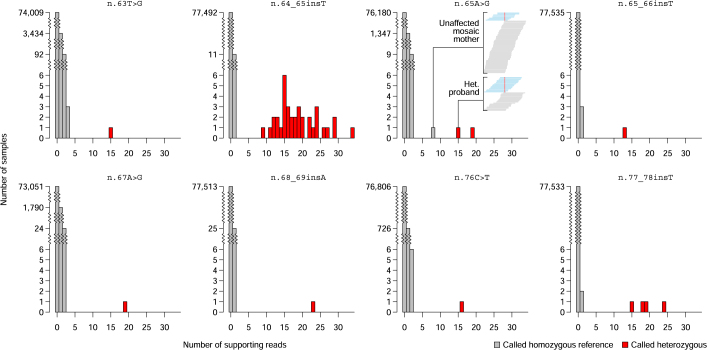
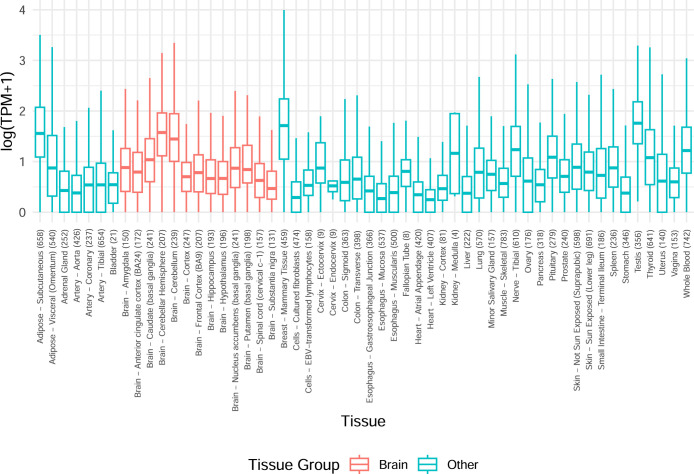
Most people with intellectual disability (ID) do not receive a molecular diagnosis following genetic testing. To identify new etiologies of ID, we performed a genetic association analysis comparing the burden of rare variants in 41,132 noncoding genes between 5,529 unrelated cases and 46,401 unrelated controls. RNU4-2, which encodes U4 small nuclear RNA, a critical component of the spliceosome, was the most strongly associated gene. We implicated de novo variants among 47 cases in two regions of RNU4-2 in the etiology of a syndrome characterized by ID, microcephaly, short stature, hypotonia, seizures and motor delay. We replicated this finding in three collections, bringing the number of unrelated cases to 73. Analysis of national genomic diagnostic data showed RNU4-2 to be a more common etiological gene for neurodevelopmental abnormality than any previously reported autosomal gene. Our findings add to the growing evidence of spliceosome dysfunction in the etiologies of neurological disorders.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Genomics England PanelApp. Available from https://panelapp.genomicsengland.co.uk (accessed on April 4, 2024), intellectual disability—microarray and sequencing (version 5.515).
MeSH terms
Substances
Grants and funding
- R01 HL161365/HL/NHLBI NIH HHS/United States
- R01HL161365/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- WT_/Wellcome Trust/United Kingdom
- R03 HD111492/HD/NICHD NIH HHS/United States
- R03HD111492/U.S. Department of Health & Human Services | NIH | Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD)
LinkOut - more resources
Full Text Sources
